
3.5: Syntheses of Prostaglandins from Cyclopentanes
- Page ID
- 285446

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)Since the all trans stereochemistry of ring substituents should be thermodynamically preferred for cyclopentane derivatives such as PGE1, a method of stereocontrol less powerful than the use of temporary bridges would seem adequate for prostaglandin synthesis. Furthermore, the availability of simple cyclopentanoid precursors including cyclopentadiene, that was used in many of the syntheses described above, led to the formulation of a simple strategy for stereocontrolled total synthesis of prostaglandins. Furthermore, such strategies are well-suited to enantioselective total synthesis (see section 3.6). Polar reactivity analysis of PGE1 as in 186 suggests dislocation of this stereochemically complex target into two fragments 187 and 188 containing only one stereocenter each.15 Thus, steric approach control might favor ddition of the vinyl nucleophile 188 from the less congested face of the cyclopentanone ring, the face opposite a substituent at position 11.

Furthermore, 1,4-additions of lithium diorganocuprates such as 189 with α,β-unsaturated ketones are especially susceptible to such SAC. However, lithium diorganocuprates were also known to displace allylic oxygen such as that at the 11-position in the cyclopentenone 187 or at the 15-position in the lower prostaglandin side chain as in the hypothetical reaction of 190 to generate 191, a useless byproduct. Should such a potentially fatal flaw preclude further consideration of a synthetic strategy? The answer depends on the value of the possible discovery that the flaw is not fatal. This leads to another rule of thumb to be added to the list began in section 1.5:

(6) Favor potentially flawed strategies only if the effort involved in further examination of the possible flaw is offset by the potentially great reward of an especially elegant and efficient synthesis.
Woodward's strategy involving intramolecular cycloaddition of 134 to generate 133 is an example of this principle not paying off. Woodward's strategy involving intramolecular alkylation of ketone 160 and his ultimate success in achieving the required skeletal connection by a modification of the strategy is an example of yet another principle of synthetic planning:
(7) Devise backup strategies, especially for risky steps.
The 9,8,12,11-circuit in 187 is dissonant. One strategy for generation of this dissonant functional array (see page 88) involves 1,4-dioxidative addition (4πs + 2πs cycloaddition) of singlet oxygen to a monosubstituted 1,3-cyclopentadiene precursor 194 to generate an endoperoxide 192 that could undergo disproportionation to the required hydroxycyclopentenone in analogy with the disproportionation of PGH to PGE.15 Alkylation of cyclopentadienide anion with bromoester 195 would produce a 5-substituted 1,3-cyclopentadiene. However, the requisite 2-substituted isomer is readily available because monoalkyl 1,3-cyclopentadienes exist at room temperature as an equilibrium mixture of mainly 1 and 2-substituted isomers that are formed from the 5-substituted isomer by [1.5] sigmatropic hydrogen migrations.

Alternatively, a simple monosubstituted cyclopentenone 193 might be converted to 187 by allylic oxidation. A route to 193 is suggested by the possibility that the C=C bond in this enone can be produced from a cyclopentanone 196 by elimination of water. If the leaving group is a hydroxyl, the presence of such functionality at the 8-position in a precursor 196 invites further dislocation to a nucleophilic upper side chain synthon 197 and a carbonyl electrophile, 1,2-cyclopentanedione. The electrophilic carboxyl functionality in 193 is latent in 197 to avoid undesired intramolecular reaction with the nucleophilic center at position 7.
It is interesting to note that the two routes to 187 outlined above involve electronically complimentary polar strategies for generating the 7-8 bond. One route exploits an upper sidechain electrophile and a cyclopentyl nucleophile (i.e. 195 and cyclopentadienide anion) while the other route exploits an upper sidechain nucleophile and a cyclopentyl electrophile (i.e. 197 and 1,2-cyclopentanedione).
Bromoester 195 was prepared from tetrahydropyran and diethyl malonate. Singlet oxygen, generated chemically, reacts with the monosubstituted cyclopentadienes 198-200 under basic conditions to deliver hydroxycyclopentenone 201 and its isomer having a hydroxyl at the 9-position and a carbonyl at the 11-position. The latter isomer was readily converted to 201 by an oxidation and reduction sequence.

A Grignard reagent synthetic equivalent of nucleophilic synthon 197 was prepared by monohydroboration of 1,7-octadiene followed by iodo-deborination and reaction of the resulting iodide 202 with magnesium. Oxidation of cyclopentanone provides 1,2-cyclopentanedione whose methyl enol ether 203 delivered cyclopentenone 204 upon reaction with 7-octenyl-magnesium iodide followed by hydrolysis of the enol ether and dehydration. Generation of an ester from the latent precursor required selective oxidative cleavage of one C=C double bond in 204. This was readily achieved by epoxidation of the more electron-rich C=C bond with peracid followed by oxidative cleavage of 205 with periodate. Methylation delivered ester 190 that was allylically brominated to provide 207 after hydrolysis of an intermediate bromide 206.
A lower side chain vinyl nucleophile is prepared by hydroalumination of (S)-1-octyne-3-ol (208) followed by iododealumination of an intermediate vinyl alane to deliver optically pure vinyl iodide 209 of correct absolute configuration. This iodide is also available by chloroacylation of acetylene with valeryl chloride followed by iododechlorination of an intermediate vinyl chloride to deliver iodoketone 211 that affords racemic 209 upon borohydride reduction. Resolution of racemic 209 can be achieved with the phenethylamine salt of the hemiphthalate derivative. The hydroxyl group in 209 must be masked prior to lithium-iodine exchange. Reaction of 209 with ethyl vinyl ether affords an α-ethoxyethyl (EE) derivative 212 that provides a divinyl cuprate 213 by metal-halogen exchange with t-butyllithium followed by addition of \(\ce{CuI}\) and \(\ce{Bu3P}\).
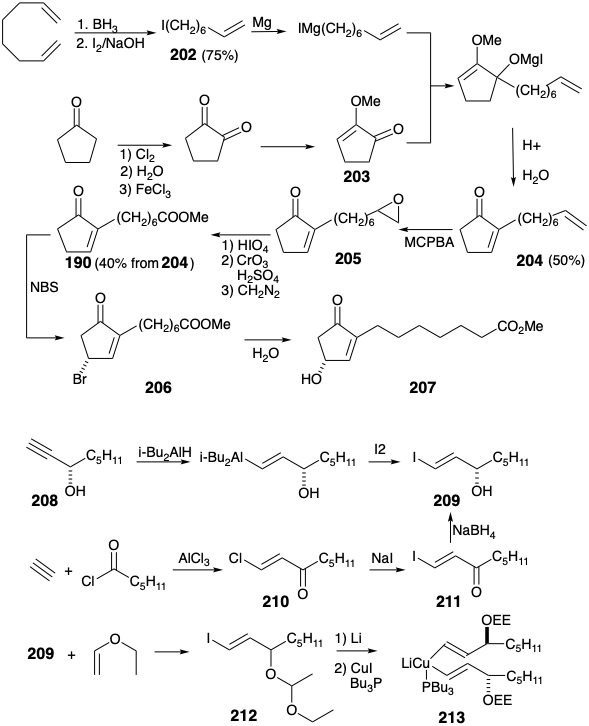
The key 1,4-addition of optically pure divinylcuprate 213 to the THP derivative 214 of racemic hydroxycyclopentenone 201, followed by removal of THP and EE protecting groups, delivers an almost 1:1 mixture of (-)-PGE1 ethyl ester 215 with the absolute stereochemistry of the natural product and its diastereomer that is epimeric at positions 8, 12, and 11. Hydrolysis of the ester to produce PGE1 could be achieved under especially mild conditions by incubation with baker's yeast. Reaction of optically pure divinyl cuprate 214 with optically pure 214 (see section 3.6) delivers (-)-PGE1 exclusively.16

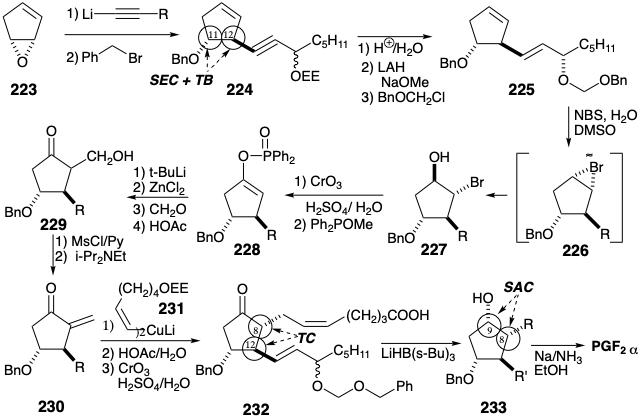
Another strategy for synthesis of prostaglandins from cyclopentane precursors17 exploits steric approach control during hydride reduction of a PGE2 derivative 216 to provide the correct configuration at the 9-position in PGF2α. Polar analysis of 216 suggests that the upper side chain can be appended by reaction of a cis vinyl nucleophile 218 with an α,β-unsaturated ketone 217. Polar analysis of 217 suggests a further dislocation to ketone enolate 219 and formaldehyde. A regioselective synthesis of the requisite enolate could be accomplished by reductive cleavage of α-bromo ketone 220. Appropriate functionalization of olefin 221 might be feasible through 1,2-dioxidative addition. That 221 might be obtained stereoselectively through regioselective nucleophilic opening of cyclopentadiene monoxide (223) by a vinyl nucleophile 222 is the reasonable consequence of an SN2 mechanism with attack at the weaker allylic C-O bond. Thus, cleavage of a temporary bridge, the epoxide, will proceed with inversion of configuration at one terminus leading to a trans relationship between the nucleophile and nucleofuge groups which become the substituents at positions 12 and 11 respectively.

The lithium acetylide from 3-(α-ethoxyethoxy)-1-octyne can serve as a terminal vinyl carbanion equivalent. Thus, reacts with epoxide 223 to afford 224 after benzylation of an intermediate alkoxide. Hydrolysis of the EE protecting group followed by trans stereoselective hydride reduction of an intermediate propargyl alcohol in the presence of methoxide followed by masking of the resulting allylic alcohol affords 225. That hydroxy bromination of 225 occurs stereo and regioselectively apparently results from a steric preference for the α-bromonium ion 226 that is attacked by water at the least sterically congested position, i. e. remote from the bulky substituent at position 12, delivering 227. That the cyclopentene C=C bond reacts in preference to the side chain C=C bond is a consequence of the electron withdrawing deactivating effect of the allylic oxygen substituent. Oxidation of 227 to the corresponding ketone followed by a Perkow reaction delivers the enol derivative 228 regiospecifically. Generation of an enolate from 228 by reaction with t-butyllithium regiospecifically activates the 8-position for nucleophilic reaction with formaldehyde delivering 229. This aldol condensation is promoted by a temporary bridge that is provided by a chelating zinc cation. Dehydration of 229 then affords enone 230 that adds a cis vinyl cuprate 231 to produce the upper side chain in 232 after selective hydrolysis of the EE protecting group and oxidation of the primary alcohol. Stereoselective, i.e. SAC, hydride reduction of 232 affords PGF2α after reductive removal of the benzyl and benzyloxymethyl ether protecting groups in 233.
Stereospecific opening of epoxides by carbon nucleophiles can be exploited to introduce both prostanoid side chains onto a cyclopentane nucleus. A remarkable strategy for the total synthesis of PGF2α from cyclopentadiene18 first simplifies the target by disconnection of the upper side chain in the usual manner at the C=C bond. The key step in the strategy involves the regioselective SN2 displacement of an electrophile at position 12 by a nucleophilic lower side chain trans vinyl carbanion synthon 222. The requisite trans relationship between the substituents at positions 11 and 12 is assured by a temporary epoxide bridge in 235 between the stereocenters at positions 11 and 12. This epoxide might be generated from the corresponding trans diol monotosylate 236. Introduction of a nucleophilic fragment of the upper side chain might also be achieved stereospecifically by an SN2 attack on an epoxide 237, a symmetrical electrophile containing a temporary bridge between the incipient 8 and 12-positions. Stereoselective generation of 237 might be achieved by steric approach control during epoxidation of a precursor cyclopentene 238. Finally, the cis relationship between the oxygen substituents in 238 can be assured by a third temporary bridge, this time between two oxygen atoms in an endoperoxide precursor 239 that is available from 1,3-cyclopentadiene by 2π + 4π cycloaddition of singlet oxygen.

Reductive cleavage of the temporary peroxide bridge in 239 delivers a cis diol. The regiocontrol during cleavage of the epoxide intermediate 242, that could be achieved with an aluminum acetylide, apparently results from a temporary bridge between nucleophile and electrophile. Thus, the hydroxy-ethyl substituent in 242 reacts with the organoalane nucleophile. The alkoxy alane then delivers the alkynyl nucleophile intramolecularly as in 243 to the desired position 12 and not position 11. The primary hydroxyl in the tetraol 238. Finally, the cis relationship between the oxygen substituents in 238 can be assured by a third temporary bridge, this time between two oxygen atoms in an endoperoxide precursor 239 that is available from 1,3-cyclopentadiene by 2π + 4π cycloaddition of singlet oxygen.

It was postulated that regiocontrol during nucleophilic attack on the epoxide intermediate 235 might be provided by a temporary bridge between the nucleophile and electrophile. Thus, the hydroxyethyl substituent in 235 would react with an organoalane nucleophile. The resulting alkoxy alane can deliver the alkynyl nucleophile intramolecularly as in 244 to the desired position 12 and not position 11. Indeed, this reaction gave the desired regioisomeric adduct 245a in 60% yield and no trace of the undesired regioisomer 245b. Further support for this mechanistic explanation is provided by the observation that silylation of 235 prior to reaction with the alane produced a regioisomeric mixture of adducts and even favored nucleophilic attack at the 11 position by 2.6:1. Also noteworthy is the fact that the bridge involving the hydroxyethyl group and the acetylide nucleophile in 244 is fused in a trans fashion with the cyclopentane ring, whereas the epoxide bridge is cis fused. Thus, while small rings prefer cis fusions, trans fusions may be unstrained and even favored thermodynamically for larger rings.
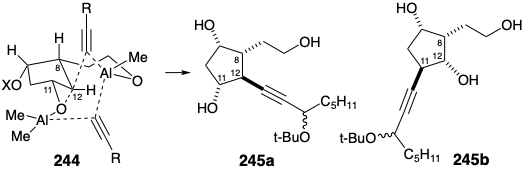
To complete the prostaglandin skeleton, the primary hydroxyl in the tetraol intermediate 242 was differentiated by tritylation. After acetylation of the remaining hydroxyls and detritylation, the resulting primary alcohol 243 was oxidized to an aldehyde before final addition of the remaining portion of the upper side chain in the usual manner.
In the foregoing strategy for synthesis of prostaglandins, polar activation that is potentially afforded by target-related functionality is not exploited for skeletal construction. Rather, electrophilicity at the 8 and 12-positions is provided by added functionality, the epoxides in 235 and 240. Economy of functionality is sacrificed in favor of incorporating temporarily bridged leaving groups that assure stereocontrol.