
30.8: Rearrangements of Cationic or Electron Deficient Nitrogen
- Page ID
- 44007
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)Since many simple nitrogen compounds are bases, they form "onium" cations when protonated. Two such cations are shown on the left (in the blue box) below. Because ammonium and iminium cations have a nitrogen valence shell electron octet, such a nitrogen atom cannot serve as a site for nucleophile bonding or a migration terminus. For a nitrogen cation to initiate rearrangement it must have an unfilled valence shell, and two such azacations are shown in the center of the diagram (pink box). An electron deficient nitrogen atom does not have to be a cation, as the nitrene example on the right (green box) demonstrates.
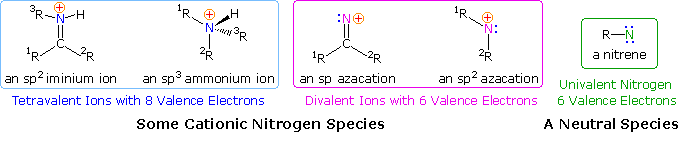
The Beckmann Rearrangement
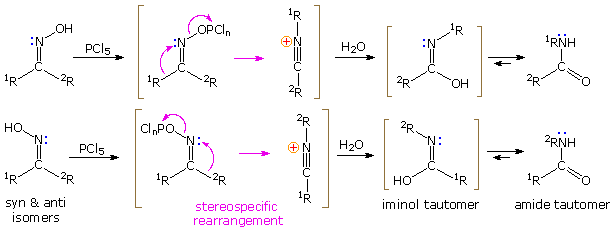
If the hydroxyl group of a ketoxime is removed by the action of strong acid or phosphorous pentachloride, followed by hydrolysis, an amide is formed. Complete removal of the derivatized hydroxyl group and its bonding electron pair would generate a divalent sp-hybridized azacation of the type depicted in the previous diagram. Were this to occur, both carbon substituents (1R & 2R) would be candidates for the subsequent 1,2-shift. In practice, however, it is always the group anti to the departing OH that migrates to nitrogen. This stereospecificity indicates that the 1,2-shift is concerted with N-O cleavage, as shown below. The resulting N-alkylated nitrilium intermediate will react with nucleophiles (e.g. water) at the electrophilic carbon atom adjacent to the "onium" nitrogen. Note that the structure drawn for this intermediate is the more favored of two resonance contributors, inasmuch as all heavy atoms have filled valence shell octets. Bonding of a nucleophile to the nitrogen atom would require expanding its valence shell to include ten electrons, or formation of an unstable dipolar species. The initial product from hydration at carbon is an iminol, which immediately tautomerizes to the more stable amide. Reactions with PCl5 probably give an iminochloride, and this in turn is hydrolyzed to the same amide.
The first example in the following group of reactions is a typical Beckmann rearrangement. The oxime from cyclohexanone has identical carbonyl substituents. and therefore exists as a single isomer. The product of the rearrangement is a lactam (a cyclic amide), which can be hydrolyzed to an omega-amino acid. This lactam serves as an important industrial precursor to nylon 6. The second example involves an oxime derivative with different carbonyl substituents, which exists as a pair of stereoisomers (syn & anti). The anti isomer rearranges by a 1,2-phenyl shift, whereas the syn isomer undergoes a 1,2-isopropyl shift. The former reaction is much faster than the latter, presumably because it proceeds by way of a relatively stable phenonium ion intermediate (structure in shaded box). Note that the picrate leaving group (2,4,6-trinitrophenolate) is a stable anion. Example #3 is another case that demonstrates the stereospecificity of the Beckmann rearrangement. The 1,2-shift of the ortho-phenol substituent is faster than that of the unsubstituted phenyl group, and the hydroxyl is ideally located to bond to the electrophilic carbon of the intermediate. Consequently, the product from the anti isomer is a benzoxazole heterocycle.
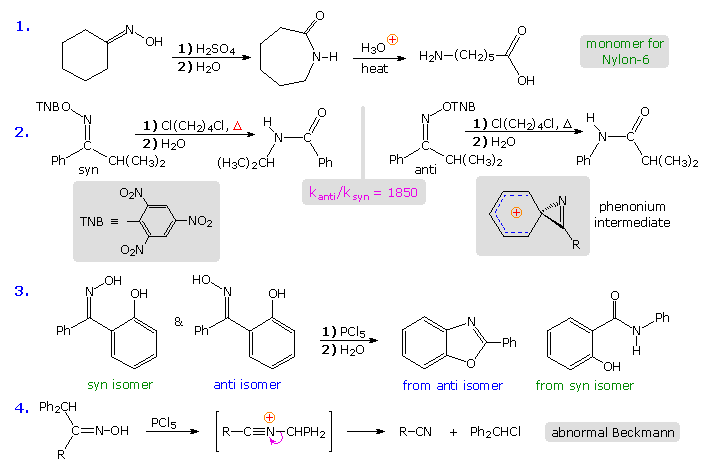
The fourth example above shows an unusual divergence in behavior that sometimes occurs when the migrating substituent fragments from the intermediate, leaving a nitrile as a stable product. This has been called an abnormal Beckmann reaction.
The rigid configuration of the phenonium cation shown above imposes a structural constraint that is nicely demonstrated by the rates of rearrangement of some fused ring bicyclic compounds. Clicking on the diagram will show the results of such a study. Oxime derivatives of phenyl ketones incorporated in six, seven and eight-membered fused rings were studied. Because of the carbon chain joining the oxime function to the ortho-carbon of the benzene ring, the phenonium ion that normally facilitates phenyl migration may be unable to assume its preferred structure (three-membered ring orthogonal to the phenyl ring). The three-carbon bridging chain for n = 6 is such a case, and rearrangement of the anti isomer is very slow. As the length of the bridging chain increases, its constraint is less severe, and the rate of rearrangement increases. The eight-membered oxime picrate hydrolyzes rapidly, producing a nine-membered lactam in high yield.
| R2C=N-NH2 + HNO2 |
Beckmann type rearrangements may also be carried out by treating hydrazones with nitrous acid, as shown on the right. As a rule, this is a less desirable procedure because pure hydrazones are more difficult to acquire, and the yield of pure product is inferior.
A direct rearrangement of ketones, thereby avoiding the necessity of preparing an derivative, is possible by a procedure known as the Schmidt rearrangement. Acid-catalyzed addition of hydrazoic acid to the carbonyl group of a ketone creates an unstable azidocarbinol that, on dehydration, produces the same triazonium cation presumably formed as an intermediate in the nitrous acid deamination of a hydrazone. Rearrangement of this species by rapid nitrogen loss then initiates a Beckmann-like rearrangement. By clicking the upper diagram a second time, two examples of the Schmidt rearrangement will be presented.
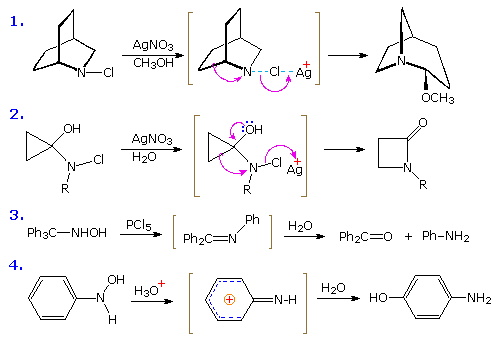
The Stieglitz Rearrangement
Examples of rearrangements to sp2 hybridized azacations are relatively rare compared with their carbon analogs. The starting materials for generating such azacations are usually chloramines or hydroxyl amines. Four examples of these transformations, sometimes called "Stieglitz rearrangements", are shown below. The first example is similar to a Wagner-Meerwein rearrangement, and the second to a pinacol rearrangement. Competitive shifts of para-substituted phenyl groups in reactions similar to example #3, demonstrate that a methoxy substituent facilitates rearrangement, whereas a nitro substituent retards it. In the last example, a conjugated azacation activates the benzene ring to nucleophilic substitution, in contrast to the usual role played by amine substituents.
Rearrangement of Acyl Nitrenes to Isocyanates
Several useful and general procedures for degrading carboxylic acid derivatives to amines all involve the rearrangement of an acyl nitrene to an isocyanate. Although the nitrogen atom of a nitrene has no formal charge, it is electron deficient and serves as a locus for 1,2-rearrangements. As illustrated in the following diagram, acyl nitrenes may be generated from different amide-like starting compounds. Once formed, acyl nitrenes quickly rearrange to relatively stable isocyanate isomers, which may be isolated or reacted with hydroxylic solvents. The most common application of this rearrangement is for the synthesis of amines. Thus, addition of water to the ketene-like isocyanates produces an unstable carbamic acid that decomposes to an amine and carbon dioxide. General procedures for obtaining the nitrene precursors are listed below the diagram.
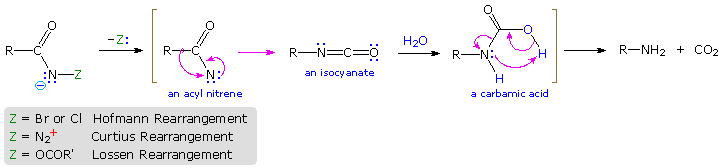
- Hofmann Route: Primary amides are converted to N-halogenated derivatives by the action of HOX or X2 in alkaline solution. Excess base generates a conjugate base of the product.
- Lossen Route: A hydroxamic acid derivative (RCONHOH) is made by reacting an ester with hydroxyl amine. The hydroxamic acid is O-acylated and then converted to its conjugate base.
- Curtius Route: An acyl azide (RCON3) is prepared in one of two ways. (i) Reaction of an acyl chloride with sodium azide, or (ii) Reaction of an ester with excess hydrazine, followed by reaction of the acylhydrazide product (RCONHNH2) with cold nitrous acid. Acyl azides decompose to isocyanates on heating.
- Schmidt Route: A variant of the Curtius procedure in which a carboxylic acid is heated with hydrazoic acid (HN3) and an acid catalyst.
Some examples of these different rearrangements are shown in the following diagram. In each case a green capitol letter (C, H, L or S) designates the type of reaction. The first three reactions illustrate the Hofmann rearrangement, which is a particularly useful method for preparing amines. The last example shows a Curtius reaction applied to a diester by way of an intermediate bis-acylhydrazide. An alternative Curtius approach to the amine product of example # 2 is also shown. The Curtius procedure is particularly useful if the isocyanate is the desired product, since the decomposition of the acyl azide intermediate can be conducted in the absence of hydroxylic solvents that would react with the isocyanate.
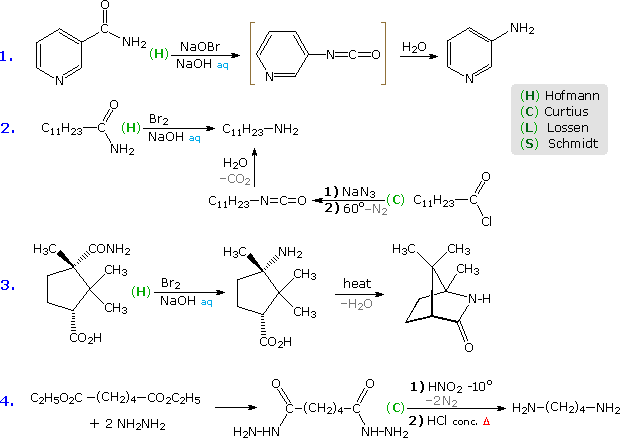
Five more examples will be displayed above by clicking on the diagram. Example # 5 shows a Schmidt reaction in which an optically active carboxylic acid is the substrate. The S-configuration of the migrating phenethyl group is retained in the amine product, confirming the intramolecular character of these rearrangements. Retention of configuration is also observed in Curtius, Lossen and Hofmann rearrangements of this kind. Reactions # 6 & # 7 are interesting cases in which water is absent during the formation and reaction of the isocyanate. The alcohol solvent in # 6 adds to the isocyanate to produce a carbamate ester, known as a urethane. Unlike the unstable carbamic acids, urethanes do not decompose and may be isolated as pure compounds. If water had been the solvent, the resulting 1º-enamine would have rearranged to an imine and hydrolyzed to an aldehyde. In the Lossen rearrangement (# 7) butyl amine adds to the isocyanate to give a substituted urea (urea is NH2CONH2).
The Hofmann rearrangement in reaction # 8 provides a novel example of the tautomerism of an acetylenic 1º-amine to a nitrile. Finally, the last example illustrates a selective Hofmann rearrangement of a bromo-imide. The reactivity of the carbonyl group para to the electron withdrawing nitro substituent is increased relative to the other imide carbonyl. Consequently, base-catalyzed hydrolysis takes place there preferentially, leaving the acyl nitrene moiety meta to the nitro function.