
Chapter 15 Solutions
- Page ID
- 1113

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
In-chapter exercises
E15.1:
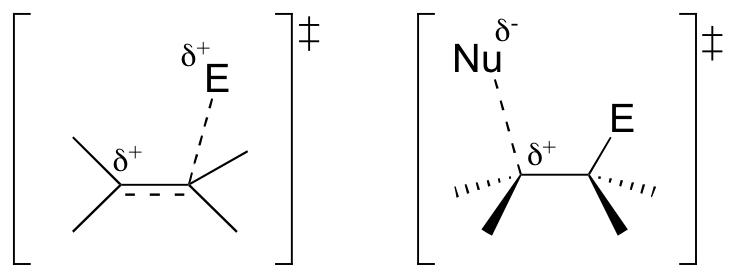
E15.2:
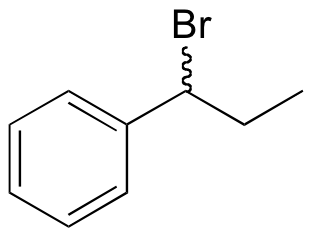
E15.3:
a)
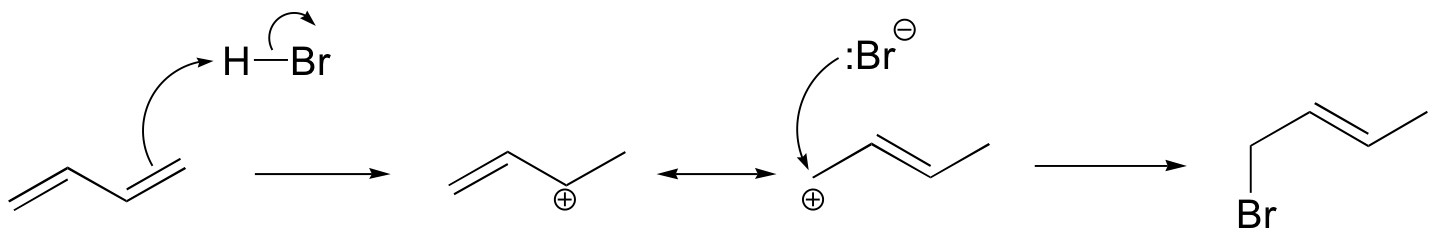
b)
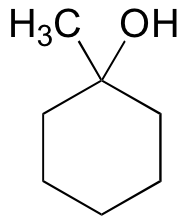
E15.4:
a) The regiochemistry of both electrophilic addition steps is governed by the relative stabilities of the carbocation intermediates. Intermediate 1 is stabilized by hyperconjugation with the neighboring methyl group, while intermediate 2 is stabilized by resonance with the bromine.
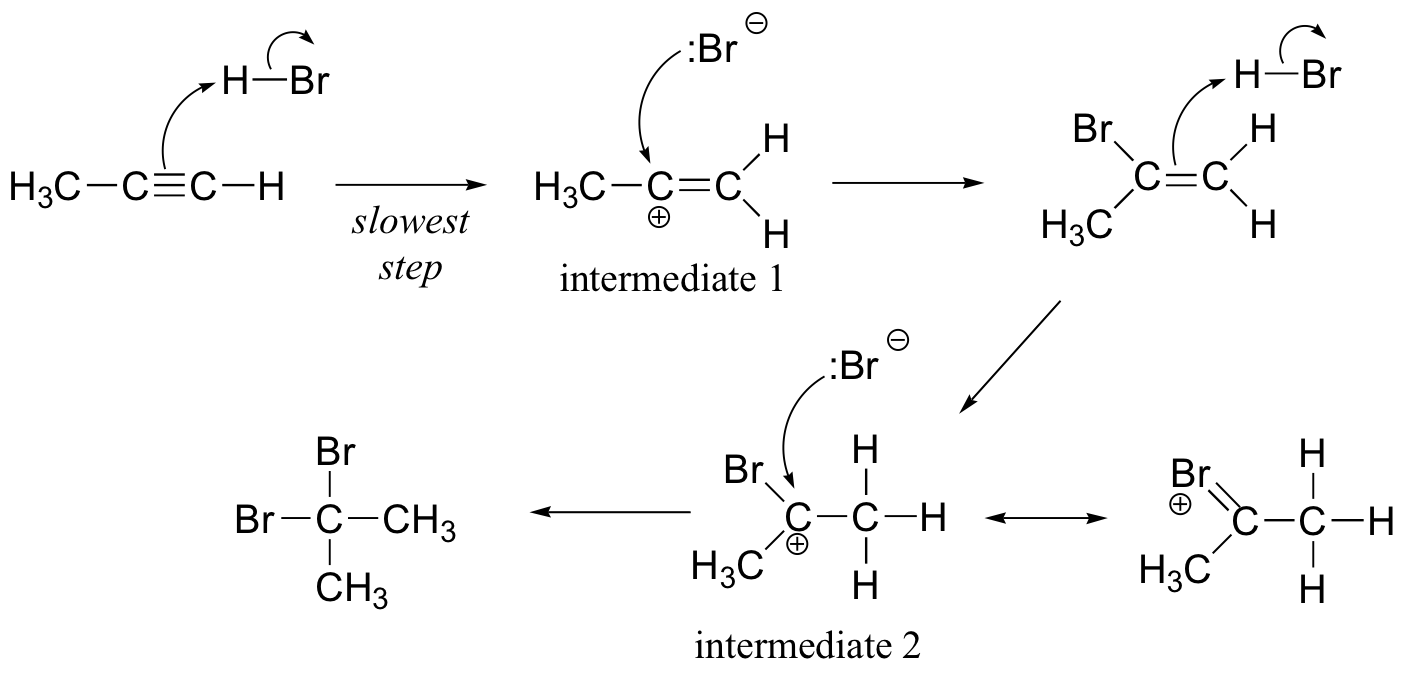
b)
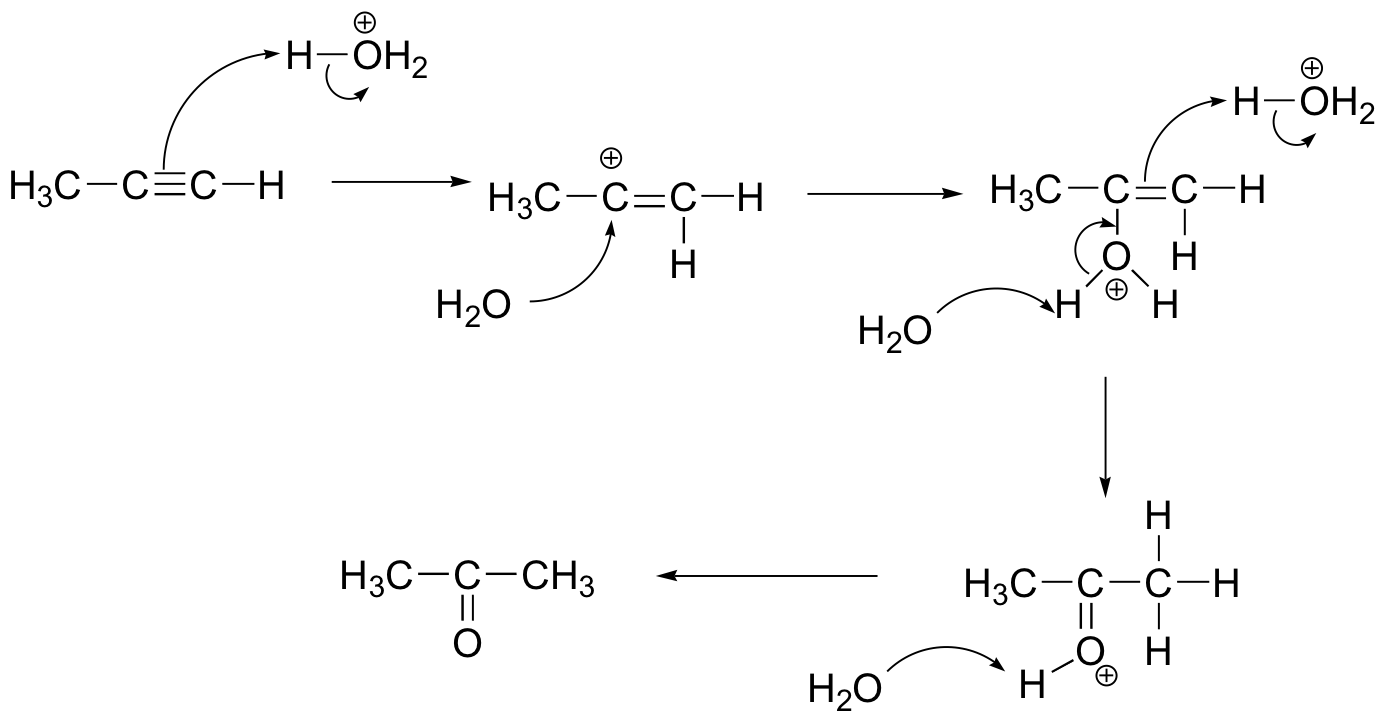
c) Alkynes are less reactive towards electrophilic addition relative to alkenes. This is because the vinylic carbocation intermediate involved is very high energy. In the case of HBr addition, for example, once the monobrominated alkene forms, the second HBr will add very rapidly.
E15.5:
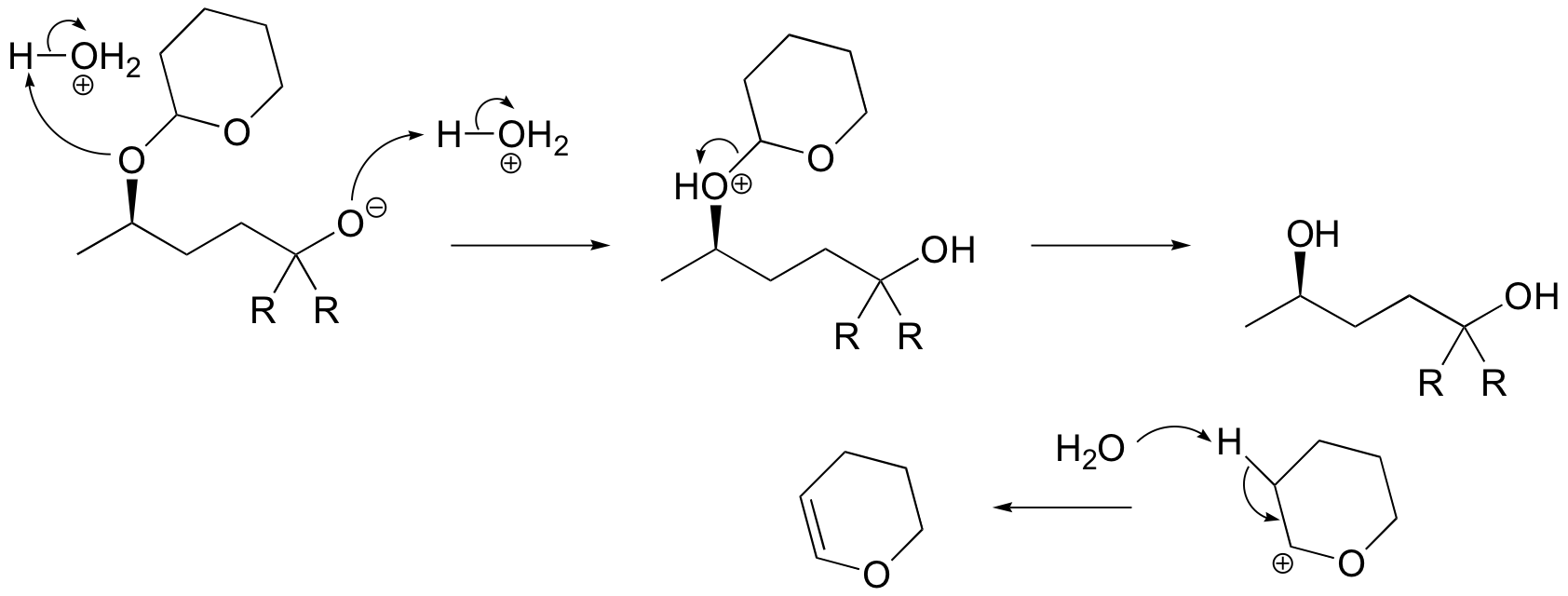
E15.6:
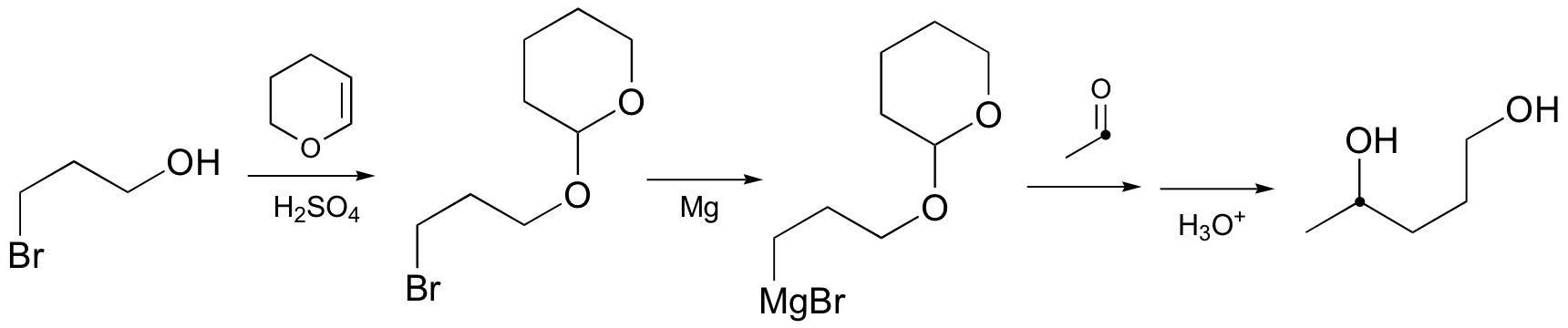
E15.7: This is an intramolecular reaction – thus there is inherently a much lower entropy component to the energy barrier. In other words, the electrophilic atom is held close to the aromatic p bonds, which makes initiation of the electrophilic attack easier.
End-of-chapter problems
P15.1:
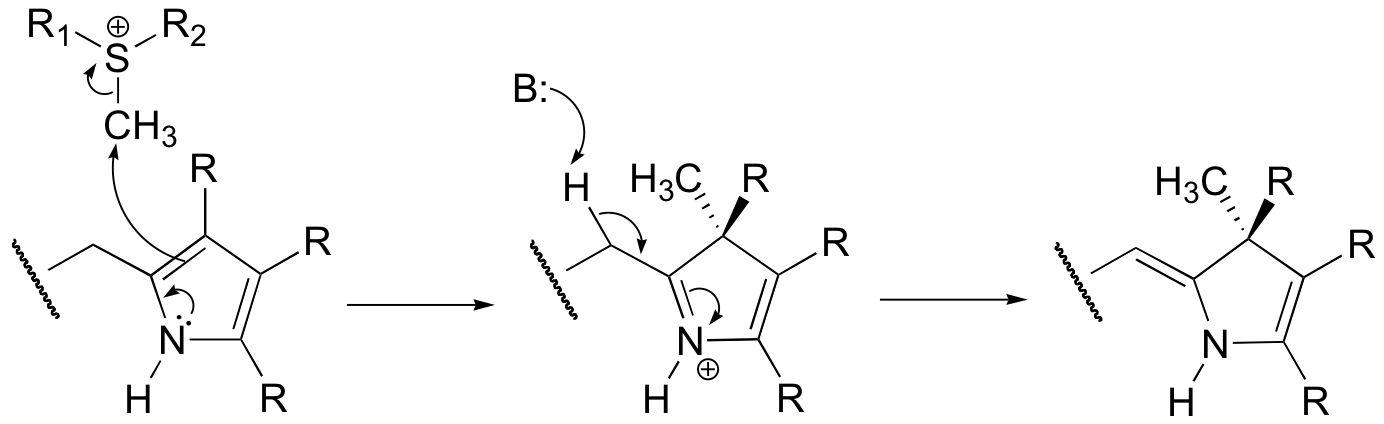
P15.2:
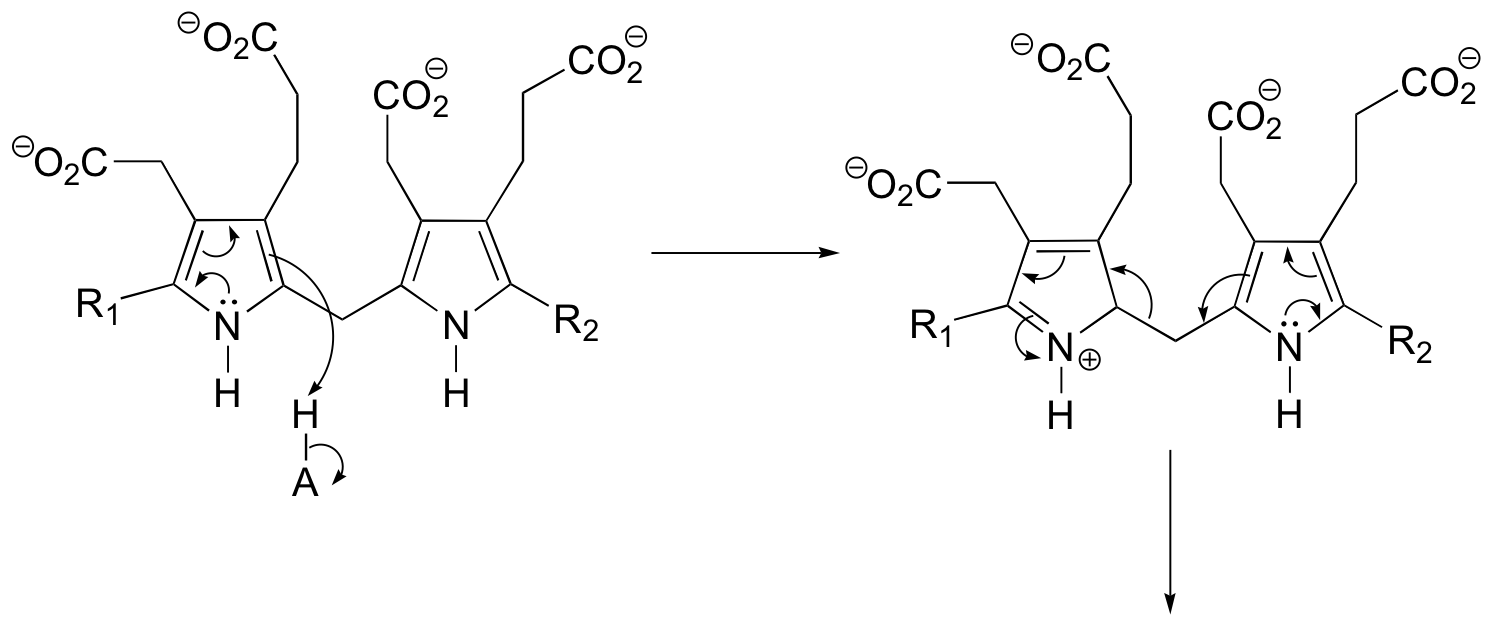
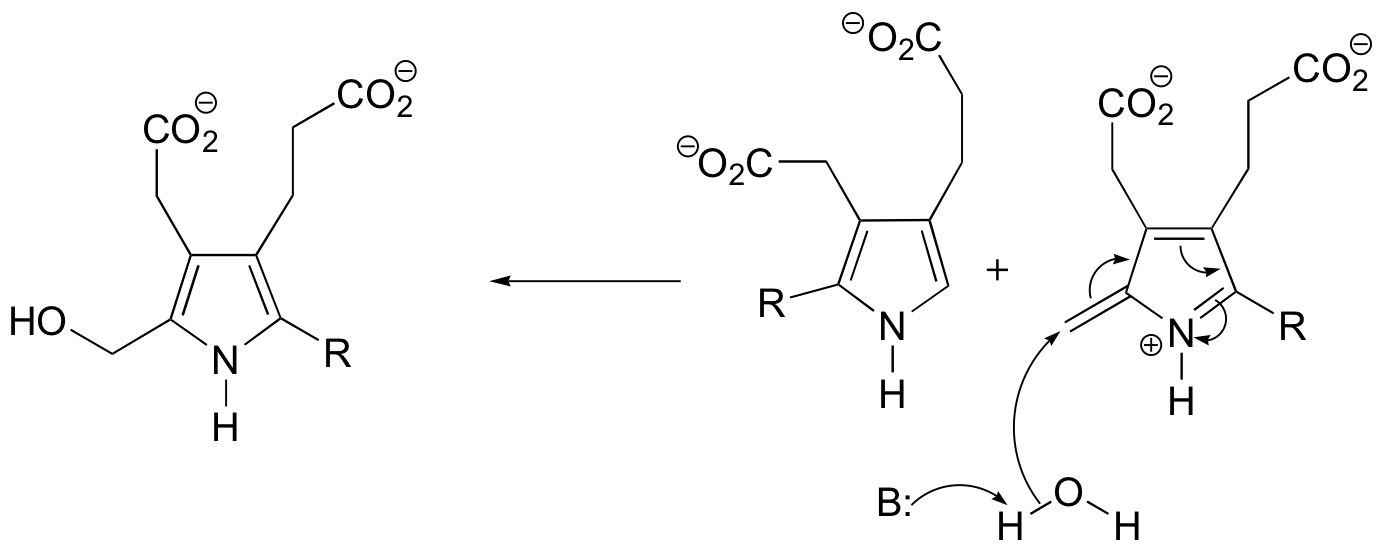
P15.3:
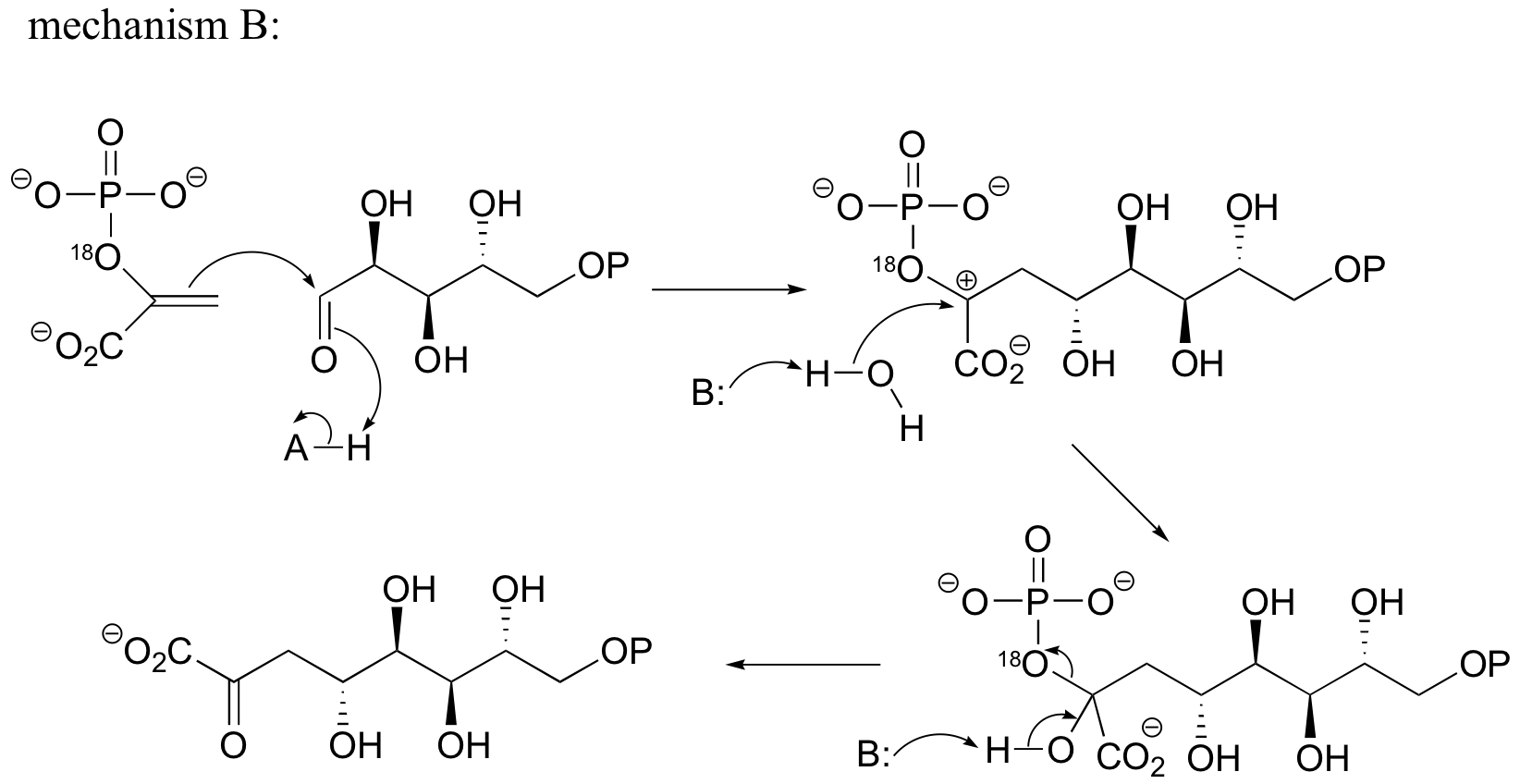
c) Mechanism B: See Biochem. Biophys. Res. Commun.1988, 157, 816.
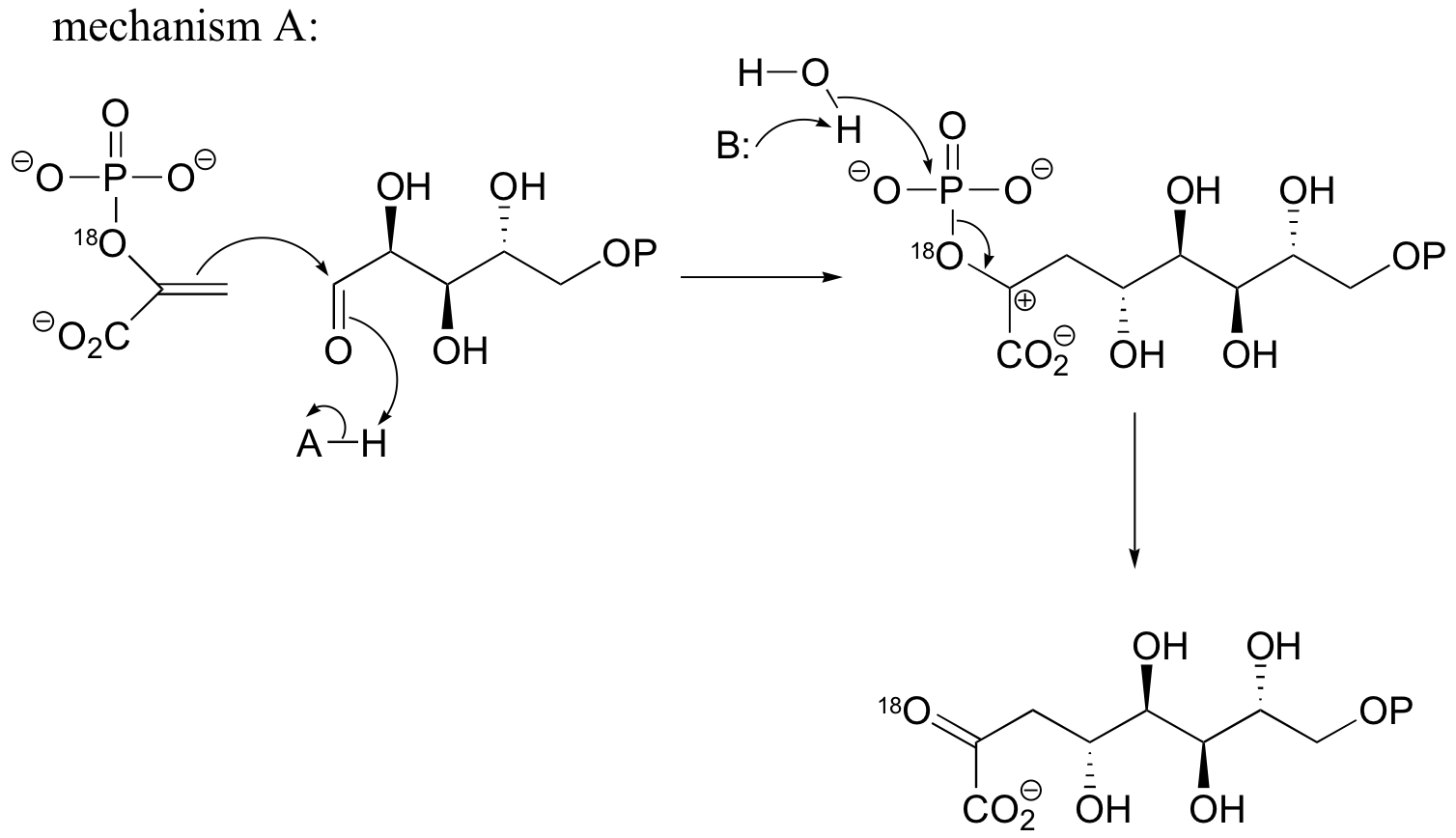
P15.4:
a)
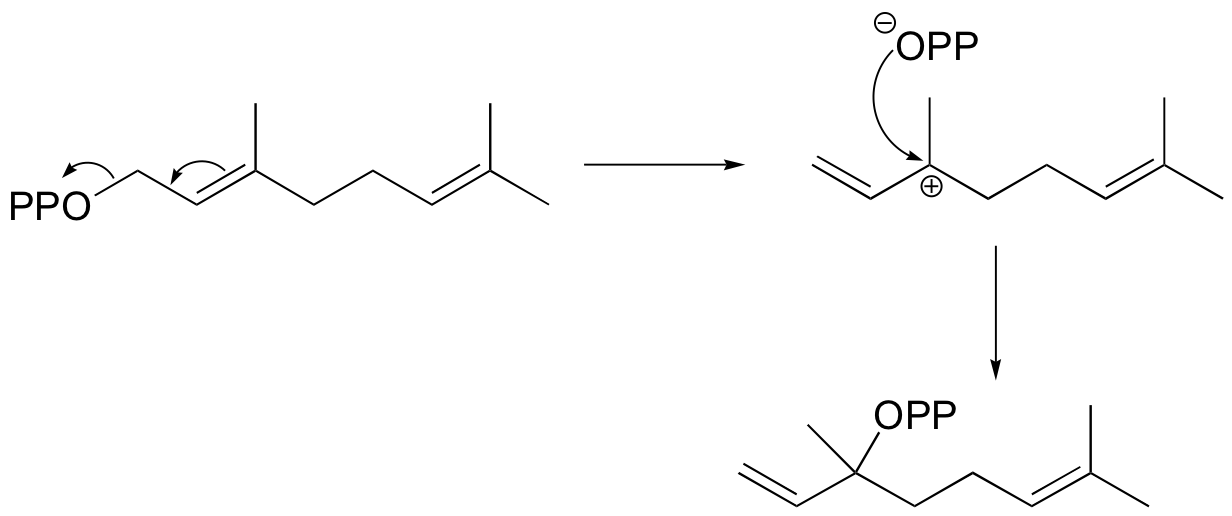
b) If the labeled substrate shown below were to undergo a concerted reaction, the label would necessarily be found on the b (outside) phosphate group of the product. If, on the other hand, the label were actually to be observed on the alpha (inside) phosphate of the product, this would rule out a concerted mechanism.
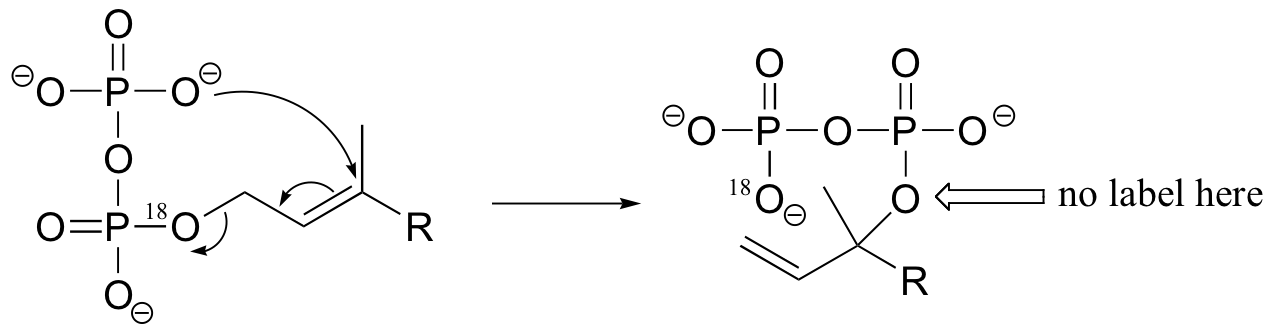
P15.5:
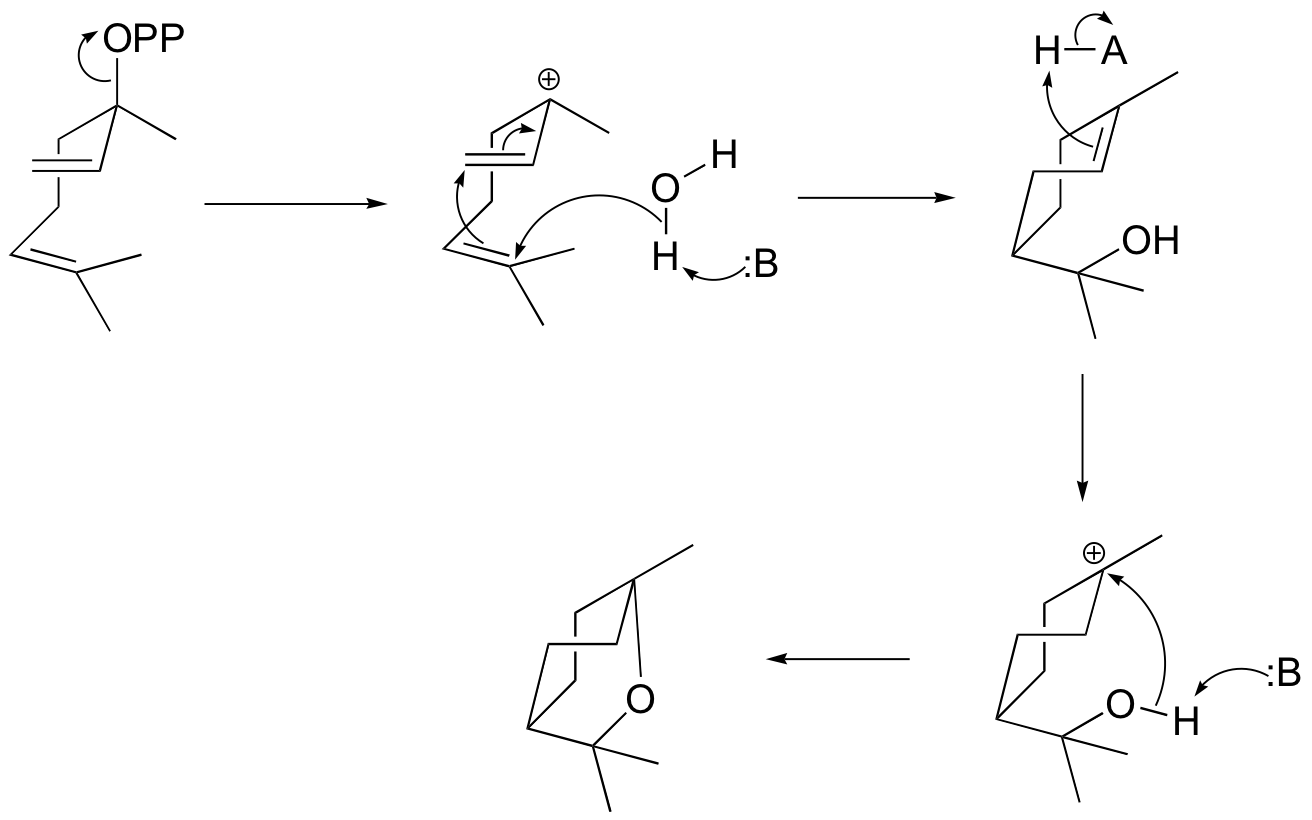
P15.6:
a)
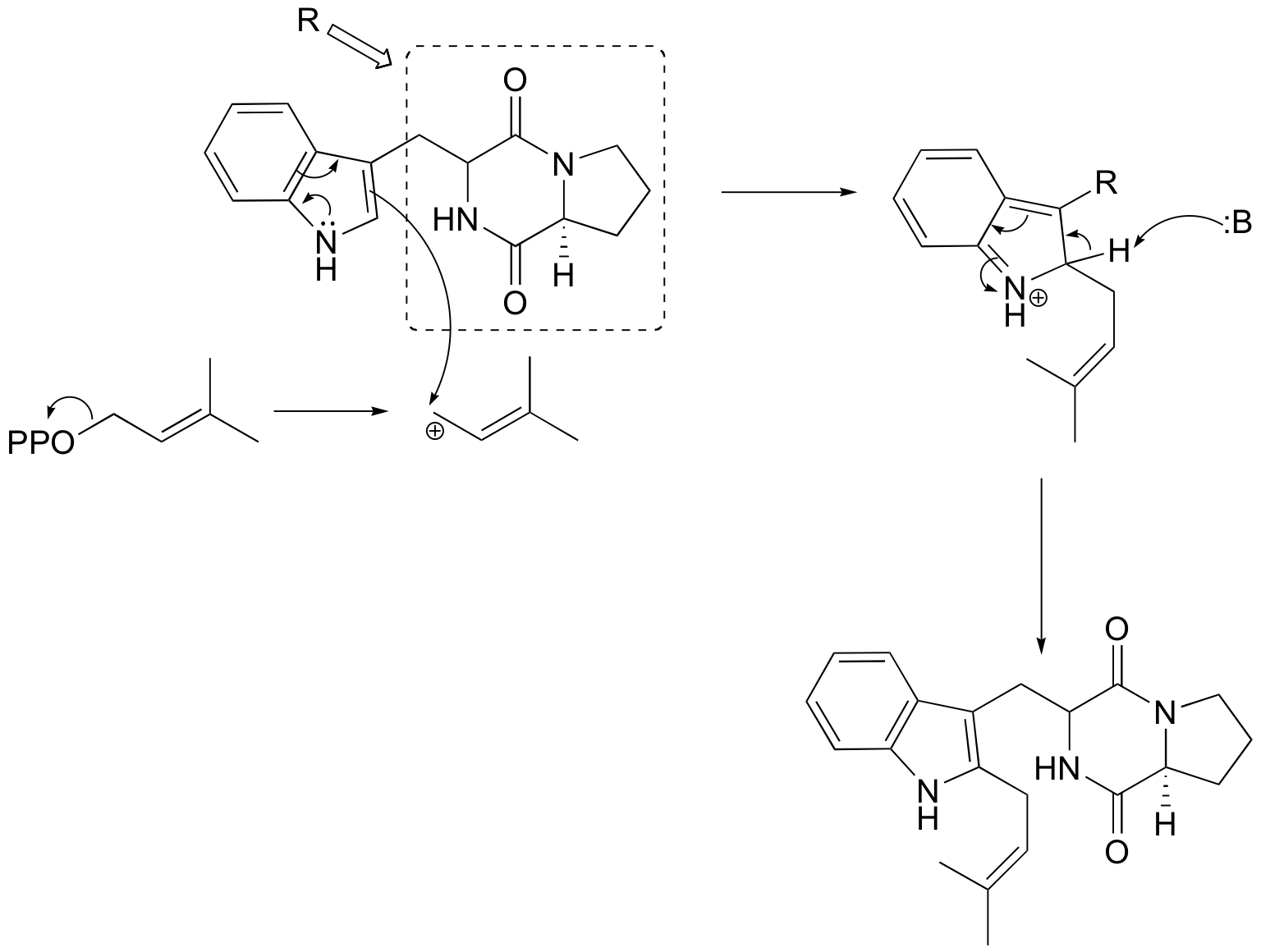
b)
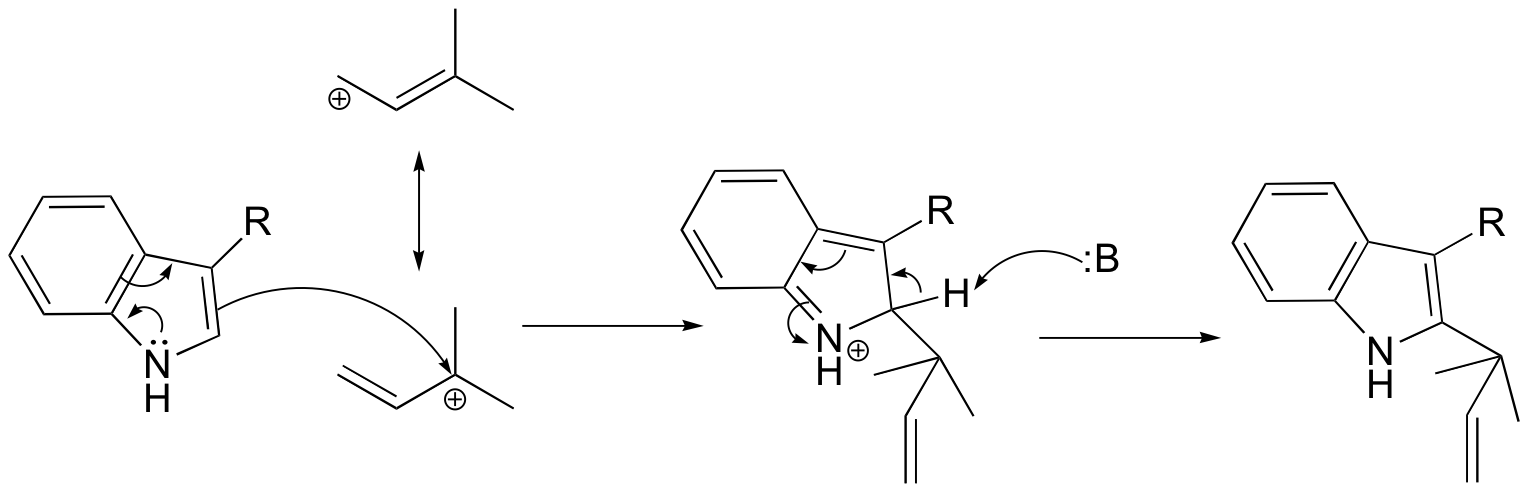
P15.7:
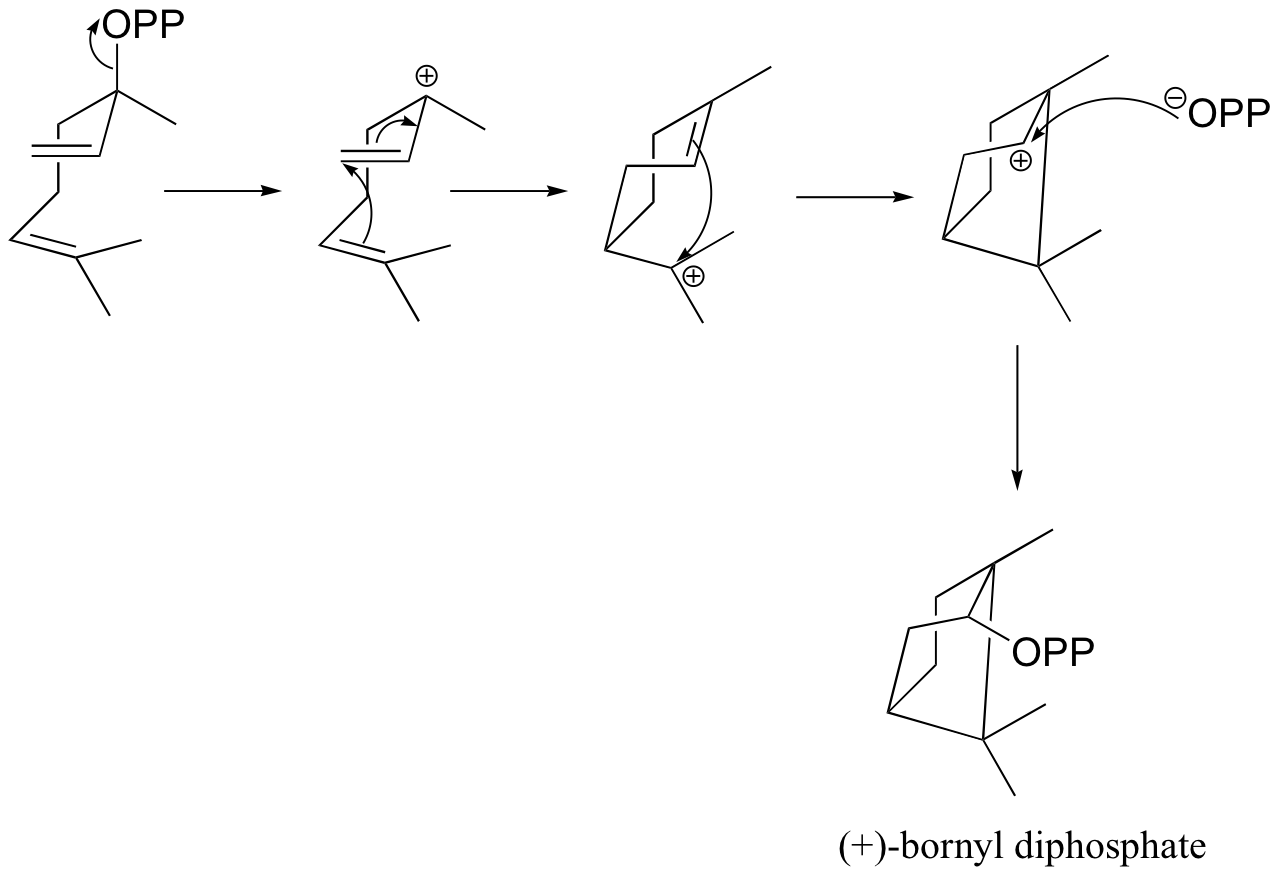
a) (+)-bornyl diphosphate
b) (+)-sabinene.
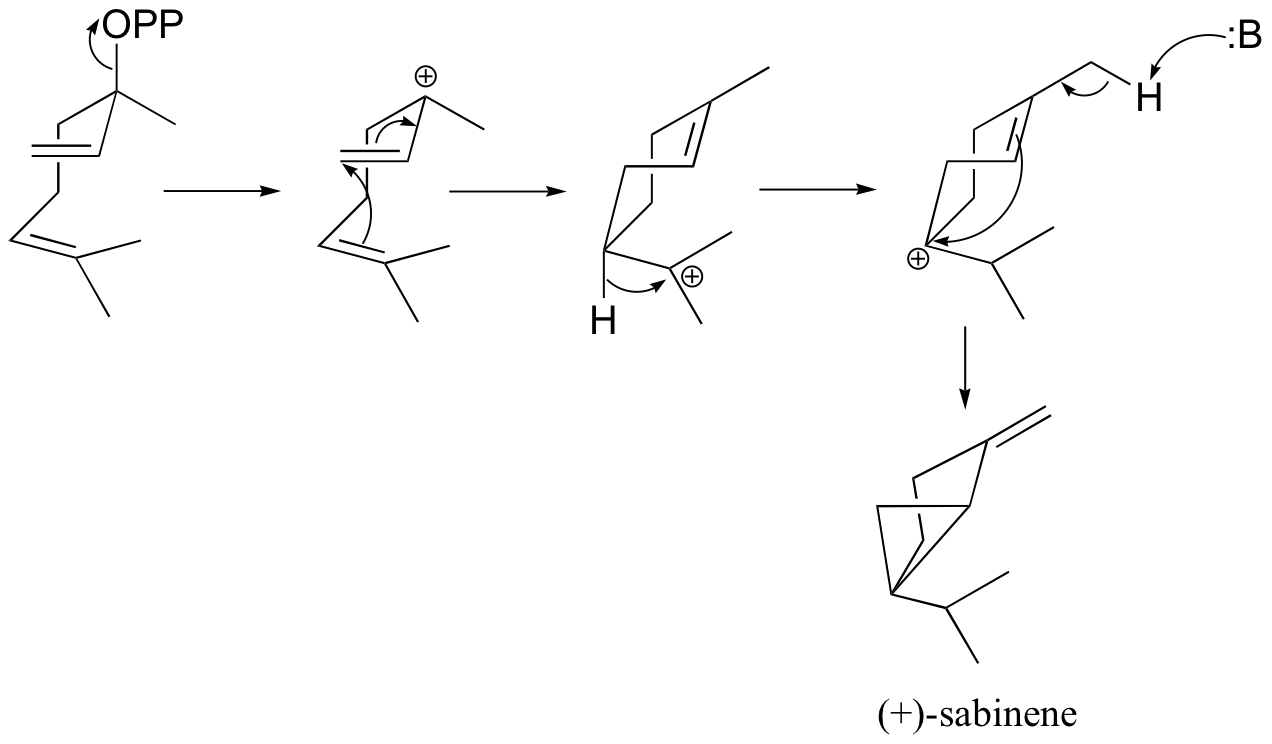
c) This is an anti-Markovnikov addition, because the secondary carbocation, rather than the tertiary carbocation, forms during the addition.
P15.8: Essentially, the cysteine is 'tricked' into acting as a nucleophile rather than as a base. (See J. Am. Chem. Soc. 2005, 127, 17433 for a complete description of the experiments referred to here).
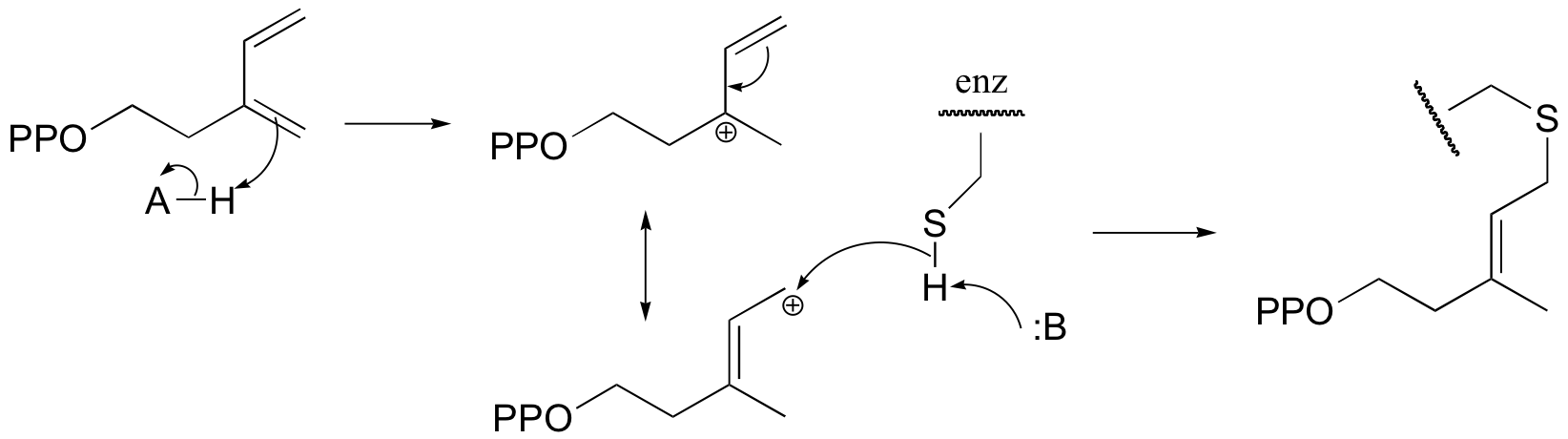
P15.9:
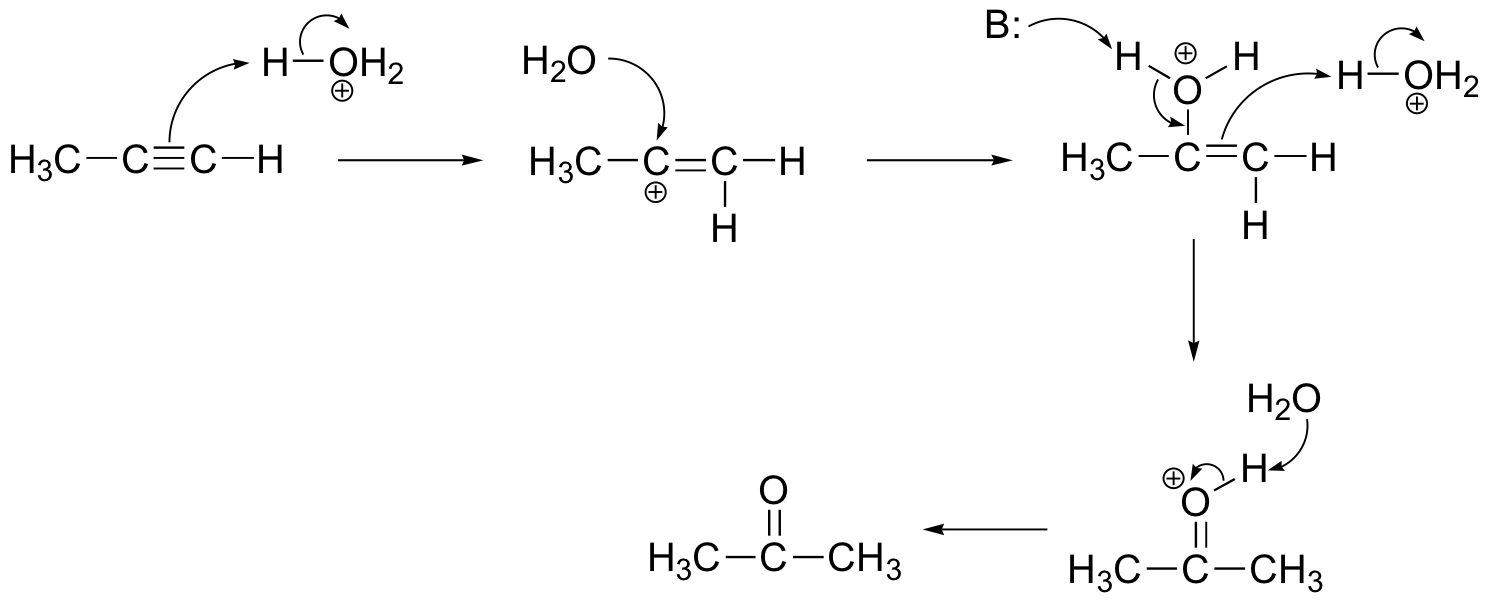
P15.10:
The NMR data shows that the main product is from anti-Markovnikov addition of HBr. This regiochemistry is due to the electron-withdrawing effect of the carbonyl group, which makes the primary carbocation intermediate more stable than the secondary carbocation.
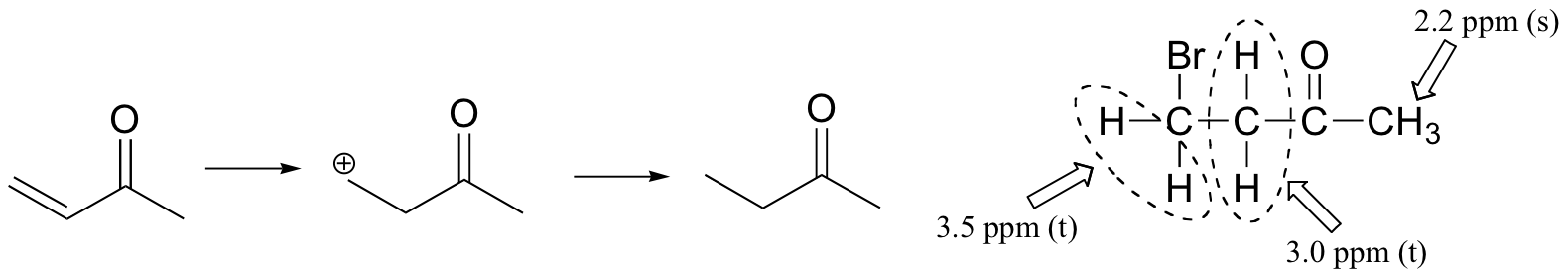
If the reaction were to proceed with Markovnikov regiochemistry, the NMR spectrum of the product would look very different:
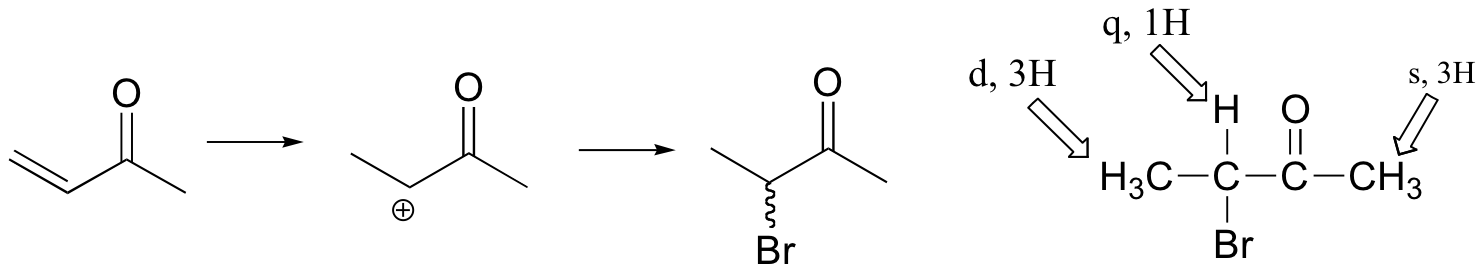
(see J. Chem. Educ. 1990, 67, 518 for more details on this experiment).
P15.11:
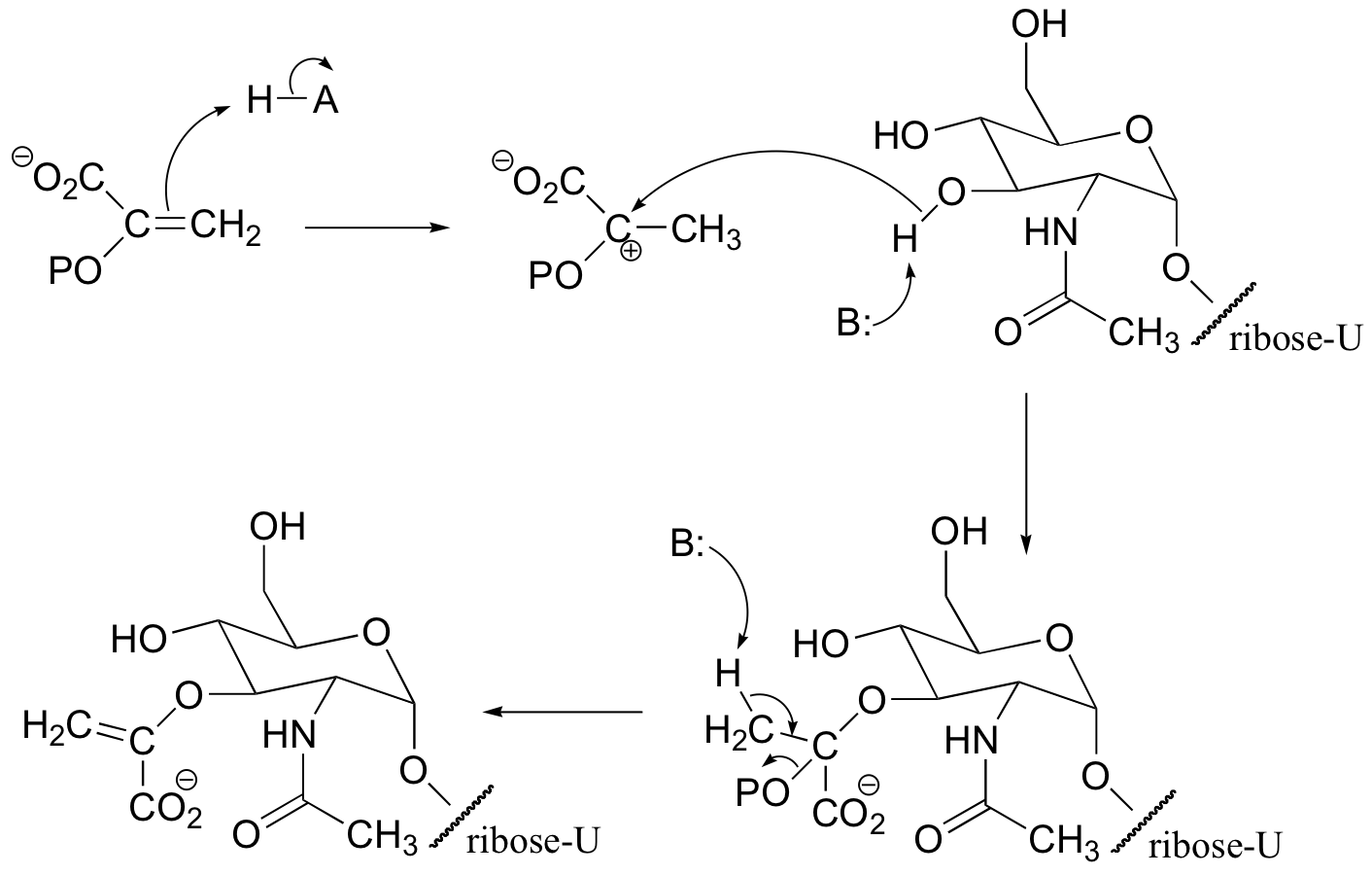
P15.12:
We would expect (and in fact we observe) alkylation to occur ortho to the phenol group, because the associated carbocation intermediate can be stabilized by resonance with the phenol oxygen.
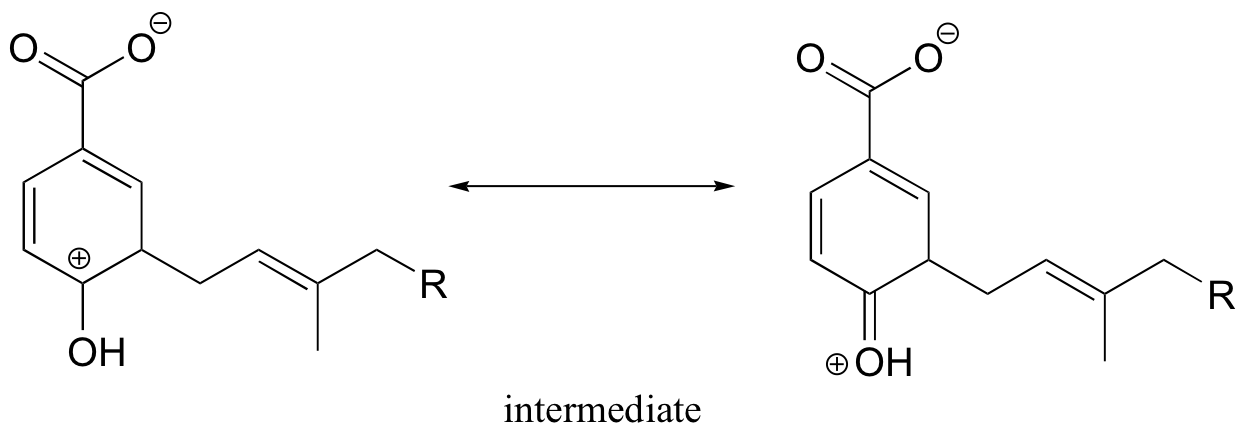
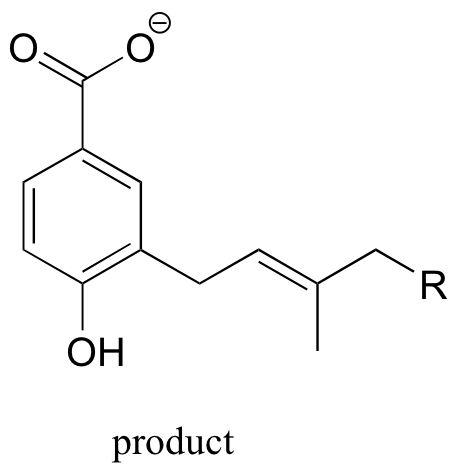
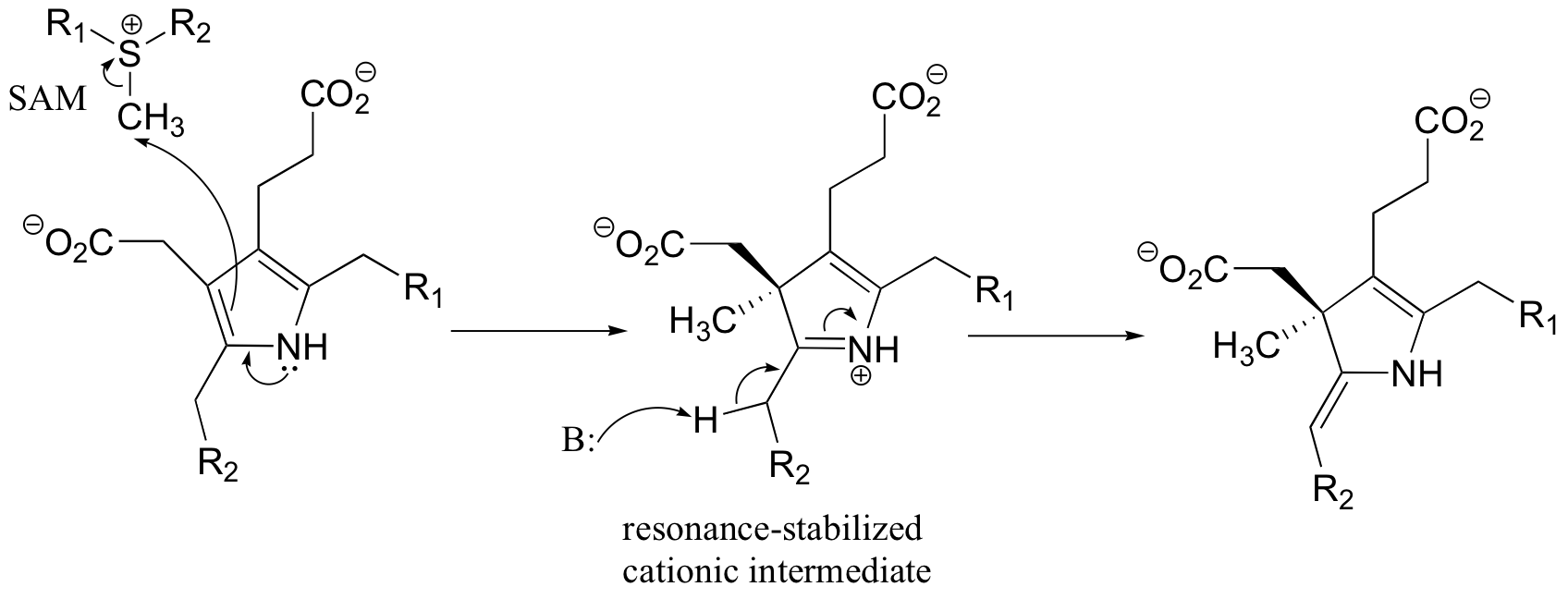
P15.13: This is a methylation reaction, so it is reasonable to expect (and in fact it is the case) that SAM is the required cofactor.
P15.14:
P15.15: See Science 1997, 277, 1815 for a detailed discussion of these reactions.
a)
b) The mechanism is same as in part a) up to the point after the hydride shift and before the methyl shift.
P15.16: This is an acyloin rearrangement (section 15.7C).
The final step of the reaction (not shown) is reduction of the aldehyde at carbon 3 to an alcohol using the reducing cofactor NADPH (this reaction type is discussed in section 16.4).
P15.17: A tyrosine in that position is able to play an additional role in stabilizing the carbocation intermediate, through a cation-pi interaction.
P15.18:
15.19:
P15.20:
a)
b)
c)
P15.21:
P15.22:
P15.23:
a)
b)
c)
d) Note that the initial product of Claisen rearrangement has lost aromaticity - but aromaticity is restored by a subsequent keto-enol tautomerization step.
Challenge problems
C15.1:
Addition can take place with either Markovnikov or anti-Markovnikov regiochemistry:
NMR data shows that it is the anti-Markovnikov product that forms, due to the electron-withdrawing effect of the ester carbonyl. (The 1H spectrum of the Markovnikov product would be expected to contain just two singlet signals.)
Peak assignments are given below. Notice that the the HR and HS protons are diastereotopic (there is a stereocenter in the molecule) and have different chemical shifts. These signals show dd splitting because HR and HS are coupled to each other, and also to the proton at 2.3 ppm.
C15.2: See Biochem. Biophys. Acta 1998, 1384, 387.
C15.3:
a) See Nature 2003, 422, 185.
b) See J. Am. Chem. Soc., 127 3577.
C15.4:
C15.5:
C15.6:
The condensation of two pyruvate molecules requires the participation of thiamine diphosphate coenzyme:
The second step is an acyloin rearangement:
C15.7: See J. Org. Chem. 2003, 68, 5433.