
1.20: Amines- Reactions
- Page ID
- 81786

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)Amines and Carbonyls - Imine Formation
Last time we looked at the behavior of amines as bases, at their involvement in hydrogen bonds, and at the ways they can be synthesized. This time, we'll continue our study of amines by examining some of their reactions.
Let's begin reviewing reactions of amines with carbonyl compounds. When we first looked at aldehydes and ketones, we learned that the characteristic pattern of many reactions of the carbonyl group begins with the formation of a bond between the carbonyl carbon and an attacking nucleophile. The nucleophile provides the electrons to form the new bond and the pi bond of the carbonyl group is broken as it "gets out of the way." The electrons move from this pi bond onto what was the carbonyl oxygen. Here's an early example in which the nucleophile is an OH- group.

This reaction step works because the OH- group is a strong nucleophile (and a strong base) very capable of using one of its unshared pairs of electrons to make a new covalent bond. If a weak nucleophile is involved, like water, the reaction needs help in the form of acid catalysis. In this pattern, the H+ begins the mechanism by making a bond with the carbonyl oxygen. The electrons which make this bond can be envisioned as coming from the carbonyl pi bond, which leaves a positive charge on the what was the carbonyl carbon.
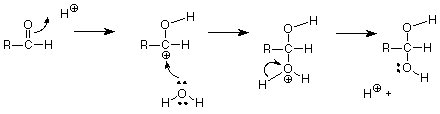
Once the pi electrons have been "gotten out of the way" by forming a new bond to the hydrogen, even a fairly weak nucleophile like water can us one of its unshared electron pairs to make a new bond to the former carbonyl carbon. The hydration reaction is completed by losing an H+, which also keeps thing tidy by replacing the H+ which was used to start the reaction.
The idea which emerges from this is that a strong nucleophile can attack directly, without help from an acid catalyst. For a weak nucleophile, an acid catalyst is needed so that the carbonyl carbon is prepared to share a pair of electrons as a new covalent bond. If we look at the mechanism of reaction between an aldehyde and an amine, we see how these factors balance. Here's the mechanism:

It is an experimental fact that this reaction -- imine formation -- is acid catalyzed. That suggests that the unshared pair of electrons on an amine nitrogen is not sufficiently nucleophilic to push the carbonyl pi electrons "out of the way" without help from an H+ which breaks that pi bond in an earlier step. Since we know that an amine (pKa of the conjugate acid ~ 10) is a weaker base than hydroxide or alkoxide ion (pKa of the conjugate acid ~ 16), it makes sense that an amine would also be a weaker nucleophile than hydroxide ion. The weaker nucleophile would be more likely to need a little help from acid catalysis.
It's also an experimental fact that if we put in too much acid, the reaction stops. How do we make sense of this? The key is to remember that an amine is a base. (Yes, sometimes it's easy to forget that something is a base if we've gotten fixated on its nucleophilic behavior, but that's our problem, not the amine's problem.) Being a base means that an amine will react with an acid to form an ammonium ion.
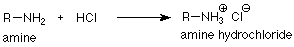
For each molecule of amine which does this, the unshared electron pair has been used to make the N-H bond and is not available to act as a nucleophile. That molecule of amine has been "benched" and is not available to react with the carbonyl compound. If this happens to all of the amine molecules (we've added too much acid) the reaction has to stop since one of its reactants is gone.
What's the best compromise? We need some amine to make the reaction go, so we want to add fewer acid molecules than there are amine molecules. We need some acid, though because it is important both to "jump start" the reaction and to catalyze the removal of the water molecule later in the mechanism. It turns out that the fastest rate happens if we control the pH so that half of the amine molecules are available to act as nucleophiles and the other half are present as the conjugate acid (ammonium salt). The ammonium ion (RNH3+) actually serves as the acid catalyst since it is the strongest acid which can co-exist with the amine. (Any stronger acid would just react with the amine to make more ammonium ion.)
Amines and Carboxylic Acid Derivatives - Amide Formation
Now let's turn our attention to the reactions of amines with carboxylic acids and their derivatives. Again, the nitrogen serves as a nucleophile in making a new bond to the carbonyl carbon. The pi bond is broken to "make room for" the nitrogen's pair of electrons. This step is just like the attack of a nitrogen nucleophile on a carbonyl carbon in an aldehyde or ketone, but what happens next is different.
The structural difference between aldehydes and ketones on one hand and carboxylic acid derivatives on the other is that a carboxylic acid derivative has a "leaving group." Leaving groups are distinguished from alkyl groups or hydrogen atoms by having an electronegative atom bonded to the carbonyl carbon. Pertinent examples include the chlorine in an acyl chloride and the -OR' group in an ester. Since the bond between one of these groups and the carbonyl carbon is polarized so that the electrons are closer to the leaving group atom than to the carbonyl carbon, it is already somewhat ionic and can cleave more easily than a carbon-carbon or carbon-hydrogen bond. This pathway is not available to aldehydes and ketones, but it dominates the reaction of carboxylic acid derivatives. The overall result is that when an amine (or any nucleophile) reacts with a carboxylic acid derivative the outcome is that the amine replaces the leaving group (a hydrogen is lost from the amine nitrogen too). The overall reaction is a substitution.
Now let's recall some examples of the reaction of amines with carboxylic acid derivatives. The details here are usually designed to overcome the fact that carboxylic acids and esters (and amides too) are less reactive than aldehydes or ketones. This is due to the fact that the "leaving group" atom in these derivatives also is electron rich (one or more unshared electron pairs) which tends to make the carbonyl carbon less "accepting" of a nucleophile's attempt to add an electron pair to it. Thus successful reactions between amines and carboxylic acid derivatives need to overcome the rather low reactivity of the carbonyl carbon in these compounds.
One very good way to do this is to put a very good leaving group in the carboxylic acid derivative. This is what is done with acyl chlorides.
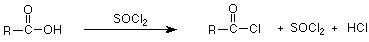
We can get a sense of how good a leaving group might be by considering how strong a base is formed when the leaving group leaves. Remember that strong bases are difficult to form and weaker bases are easier to form. Chloride ion is the conjugate base of HCl, a very strong acid, so it is a very weak base and a very good leaving group.
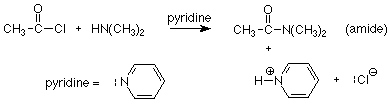
Once we have the acyl chloride with its very good leaving group, we can use a moderately effective nucleophile like an amine to get a satisfactory method for making amides.
How about reactions between amines and esters. Here, the reaction is accelerated by heating it moderately. Notice that a stronger base (amine) is used up and a weaker base (alcohol) is produced. Notice also that before the alcohol (leaving group) portion of the ester departs, it picks up an H+ so that it can leave as the weak base alcohol (R'OH) rather than as the strong base alkoxide ion (R'O-). Again, weaker bases make better leaving groups.

In the case of making amides from carboxylic acids, the difficulty comes because the carboxylic acid is a stronger acid (pKa ~5) than the ammonium salt (pKa ~10). The result is that there is very little amine and carboxylic acid at equilibrium. so there is very little nucleophile present. Also, the O- in the carboxylic acid is a very poor leaving group. This reaction doesn't look promising at all, but it can be made to work by heating the ammonium salt strongly.
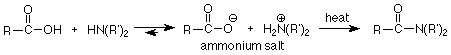
You may have noticed that we haven't tried acid catalysis of any of these reactions between amines and carboxylic acid derivatives. That's because any acid we add will react with the amine, so the strongest acid we can have in the reaction is the conjugate acid of the amine. That isn't a strong enough acid to "jump start" the lower reactivity carbonyl group of a carboxylic acid derivative.
Nitrosation - Nitroso Compounds and Diazonium Salts
Now to a reaction we haven't seen before. We will look at nitrosation because it follows on fairly naturally after the reactions of amines with carbonyl groups. Nitrosation reaction mechanisms begin with addition of a strong acid to sodium nitrite (NaNO2). Nitrous acid is formed, but it reacts further with acid to make water and the nitrosyl cation.
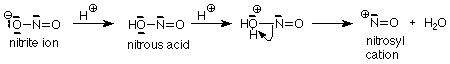
The nitrosyl cation is electron deficient. Its nitrogen has only three pairs of electrons in the valence shell, so it is a very good electrophile, very susceptible to attack by a nucleophile. When the nucleophile is a secondary amine, the product (after loss of an H+ from the amine nitrogen) is called an N-nitrosoamine.
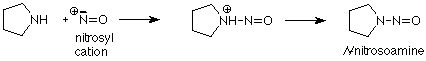
Some of these N-nitrosoamines have been shown to be carcinogenic in animals, so there is concern regarding the possibility that they may be formed when sodium nitrite (added to some meats to prevent botulism) reacts with acid in the stomach and amines present in the body. The beneficial effect of sodium nitrite in preventing botulism poisoning must be weighed against the potential hazard of N-nitrosoamine carcinogenesis. As with many dietary hazards, the longer term effects are difficult to determine.
When the amine is primary, its reaction takes a different course. We will look at an example where the R group is the phenyl group (a benzene ring), since that is the most important application of this reaction.
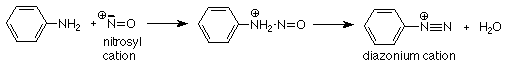
The aromatic diazonium ions produced by this reaction are stable enough to persist in a cold acidic aqueous solution. They are important as synthetic intermediates in the preparation of a variety of aromatic compounds, including dyes and photographic chemicals. We'll take a longer look at what we can do with these compounds when we study aromatic chemistry in a few weeks. In the meantime, keep this reaction in mind.