
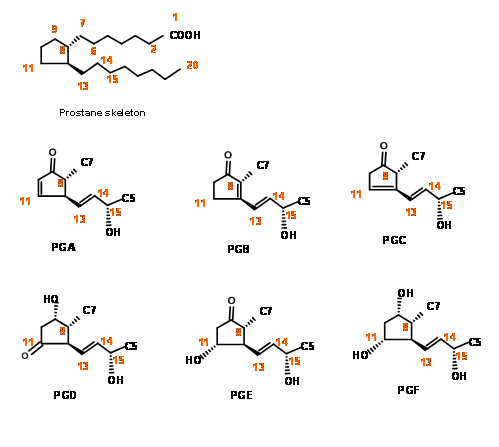
10: Strategies in Prostaglandins Synthesis
- Page ID
- 21850

Chemical Synthesis of Prostaglandins witnessed phenomenal activity during the 1960’s and 70’s. During this period, organic chemistry saw intensive development in ‘disconnection’ and ‘Logic’ as primary tools for synthesis. This period also saw development of several new reagents for stereoselective synthesis. The complexity of the structure of PG skeleton posed a great challenge for synthesis. The fact that molecules belonging to this family held great potential as drug candidates but were available only in minute quantities from natural sources was the main reason for the intense activity in the chemical synthesis and skeletal modifications for SAR studies. The numbering system on the skeleton and the main structural features of this family of molecules could be seen in Figure 10.1
Placing a keto- group at C9 is a delicate operation because such β- hydroxy ketones would readily undergo dehydration to give a PGA skeleton. On a five membered ring, PGA system could again undergo ready isomerisation to PGB, a stable ring system, probably via a PGC skeleton. On reduction with sodium borohydride, the keto- group at C9 is reduced to a mixture 9α- and the unnatural 9β- epimers. The natural PGF skeleton has an 9α- configuration for the –OH group. In the natural PGF skeleton we have four asymmetric centers on a five membered ring. As you know well, a five membered ring is conformationally very flexible. Hence setting up precise stereochemistry on this ring posed a great challenge during the 60’s. There could be three main strategies for the construction of five membered ring systems (Fig 10.2).
- An open chain could be cyclized to a ring.
- A suitable cycloalkane ring could be ring expanded or contracted to a five membered ring.
- A suitable bicyclo[l,m,n]ring system could be opened to give a five membered ring.
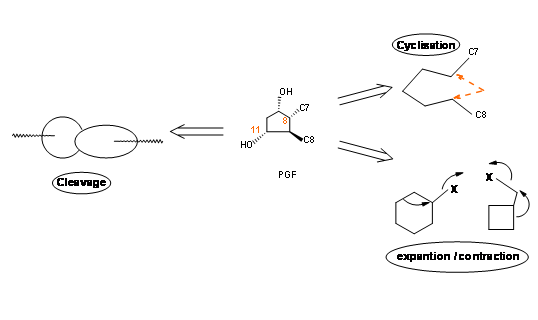
Let us look at a few outstanding synthetic efforts that successfully met these challenges in Prostaglandin chemistry.
Cyclization of open chain precursors
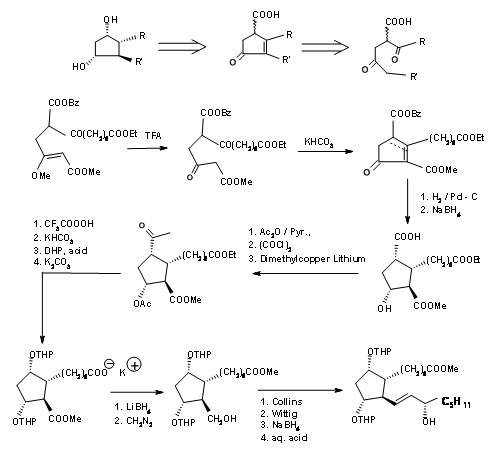
Setting up a series of asymmetric centers on an open chain is as great a challenge as setting them up on a five membered ring. Corey’s early attempt in 1968 was to design a suitable six membered ring, open up the ring to a chain and then cyclize the chain regiospecifically to the required five membered ring ( J. Am. Chem. Soc., 90, 3245 (1968)). Their retroanalysis and synthesis are shown in Figure 10.3. The first DA reaction set two crucial stereocentres that guide the remaining stereopoints on the five membered ring formed by aldol reaction. This synthesis is a good example for convergent synthesis. The starting materials come by two different routes shown in Figure 10.4.
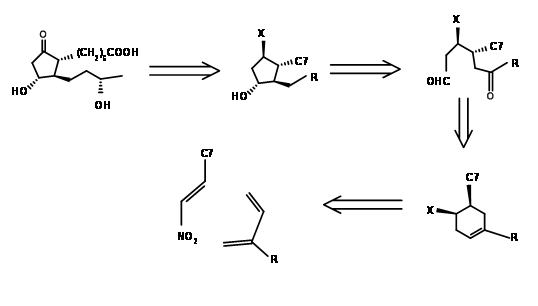
The Kojima’s disconnection of the cyclopentane ring at C8 – C12 bond provided an open chain. Careful planning of the functional groups on the proposed chain enabled his group to plan the stereocentres in a more direct way. Their retroanalysis and synthetic route are shown in Figure 10.5.
Cyclopentane ring precursor
The difficult problem of setting up stereocentres on a five membered ring was solved elegantly by Corey et.al.,(Tet. Lett., 311 (1970)) (Fig 10.6). Observe the artistic precision with which the substituents are woven into a five membered ring with the aid of iodolactonisation and epoxide ring formation reactions. An example for mastery in the Art of Synthesis.
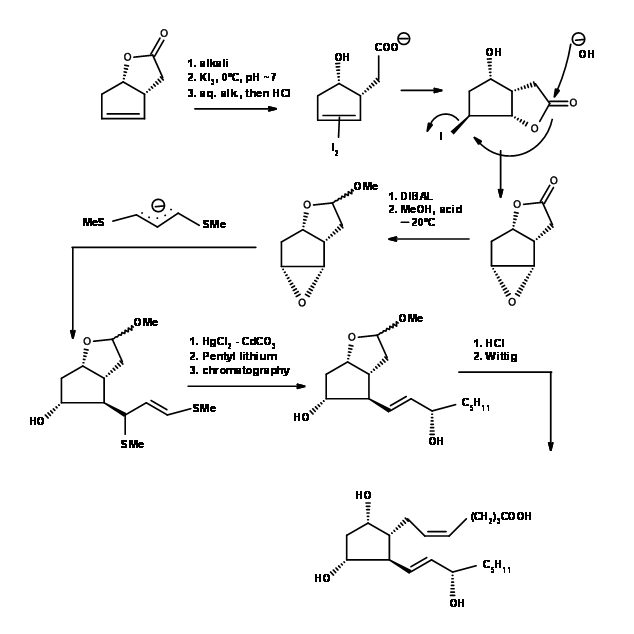
F.S. Alvarez et.al., took advantage of the steric constraints due to eclipsing strains in five membered rings and wove three asymmetric centers in a row on a five membered ring (J. Am. Chem. Soc., 94, 7823 (1972)) (Fig 10.6).
Cyclohexane ring Precursor
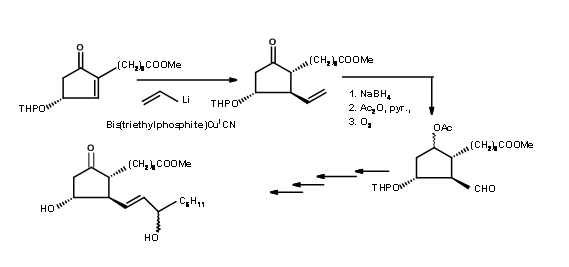
Starting from all cis-cyclohexan1,3,5-triol, Woodward’s school demonstrated their excellence in the Art of Organic Synthesis. The first step is the differential protection with glyoxalic acid (Fig 10.8). Note an interesting architectural design aspect. This chain of two carbon protecting group is eventually incorporated into the main structure. Note that this also accomplishes another very difficult task viz., placing a reactive 2 carbon chain into the crowded concave phase. The solvolysis of the mesylate is assisted by the neighbouring olefin. Note the elegant planning of the ring contraction step.
In 1973, Corey revealed a synthesis (Tet. Lett., 309 (1973)) based on six membered ring involving a ring expansion and a ring contraction to achieve the goal (Fig 10.9).
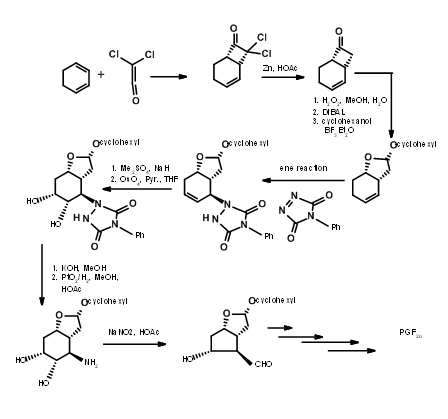
Bicycloalkane Approaches
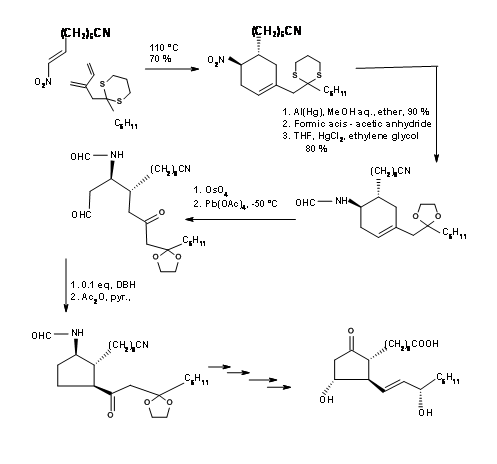
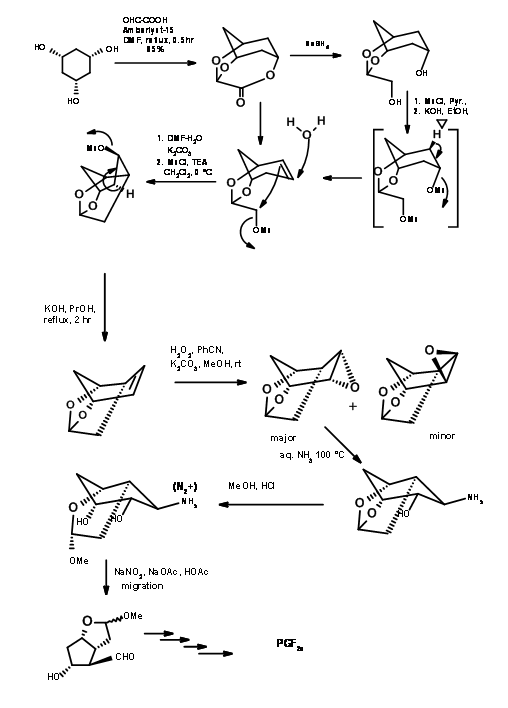
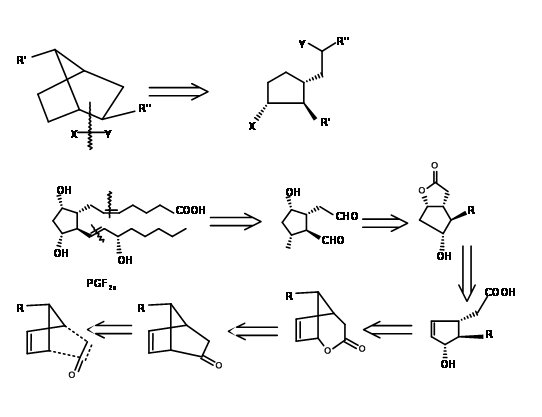
There are several syntheses based on such strategy to derive the stereochemical advantage of a bicycloalkane ring system. Here we shall discuss the Bicycloheptane strategy of Corey (J. Am. Chem. Soc., 91, 5675 (1969): Ann. N.Y. Acad. Sci., 180, 24 (1971)). This route provides entry to all natural and unnatural PGs. It provides facility for separation of enantiomers at a very early stage using Amphetamine salt procedure on the first chiral acid-alcohol. This scheme has been scaled up to multigram-scales. Corey’s retroanalysis is shown in Figure 10.10. This retroanalysis led to DA reaction.
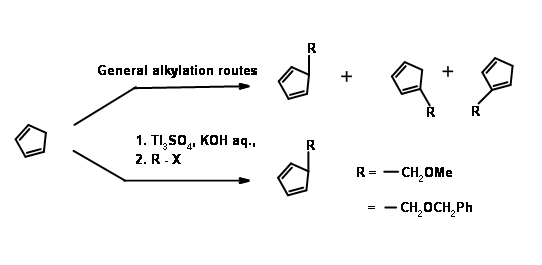
The first problem was the synthesis of a cyclopentadiene with the alkyl group at the methylene carbon. Anion routes for alkylation of cyclopentadiene mostly led to a mixture of cyclopentadienes, due to easy isomerisation of the olefin. The problem was solved by alkylation with thallium anion (Fig 10.11). The desired cyclopentadiene was obtained in about 97% yield. The next challenge was in the DA reaction.
Ketenes do not undergo Diels-Alder cycloaddition. They give cyclobutane products even with dienes. Using a ‘masked ketene precursor’ this problem was solved as shown in Figure 10.12. The bicyclo[2.2.1]heptane thus formed underwent Baeyer-Villiger oxidation as expected. Saponification of the lactone gave a five membered ring with three asymmetric centers in place. The fourth center was generated through an iodolactonisation reaction.
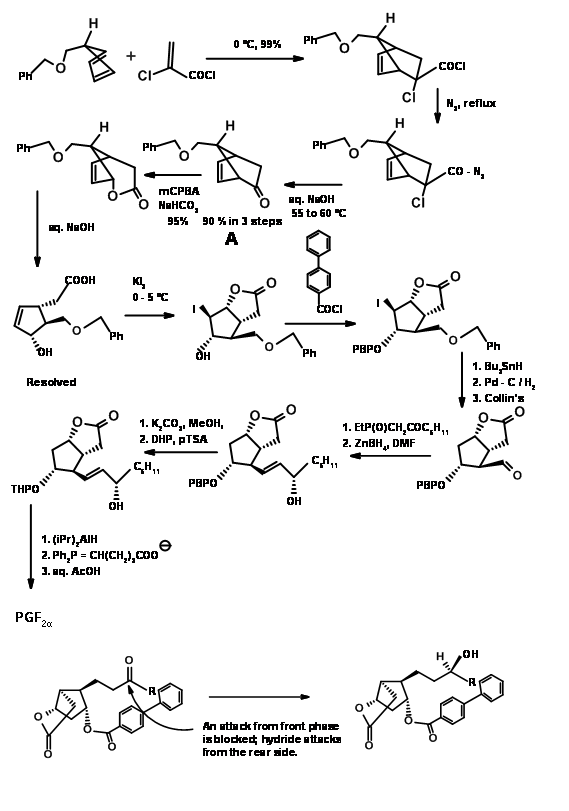
Note the stereochemistry of this iodolactonisation step. After removal of the halogen, the –OH at C11 was protected as p-phenylbenzoyl ester. This ester not only gave a crystalline derivative for purification (a useful feature during large scale reactions) but also served as a shielding agent to induce C15-(S)– OH at a later stage. Hydrogenolysis of the C13 – OR to – OH followed by Collins oxidation gave the C13 aldehyde suitable for a Wittig reaction. The α,β-unsaturated ketone was reduced selectively to C15-(S)– OH using Zinc borohydride. This selectivity has been attributed the shielding effect of the bulky aroyl group from the front side, allowing the hydride to enter stereoselectively. Reduction of the ester group with trialkylaluminium hydride gave the hemiacetal for the final Wittig. Note the differential protections used in this synthesis at different stages.