5.6: Dihydroxylation, Aminohydroxylation and Aziridination Reactions
- Page ID
- 170167

5.6.1 Dihydroxylation Reaction
In 1980, the first attempt for enantioselective cis -dihydroxylation of alkenes with osmium tetroxide was appeared. Subsequent continuous efforts led to improve the reaction yield and enantioselectivity in the presence of osmium-cinchona alkaloid complexes (Scheme \(\PageIndex{1}\)). The reactions can be performed at ambient conditions in liquid-liquid biphase system having water and t-BuOH employing secondary oxidant such as K3Fe(CN)6 to afford the target 1,2- cis diols with high enantioselectivity. Please see Module I, Reagents and Organic Reactions, for the mechanism.
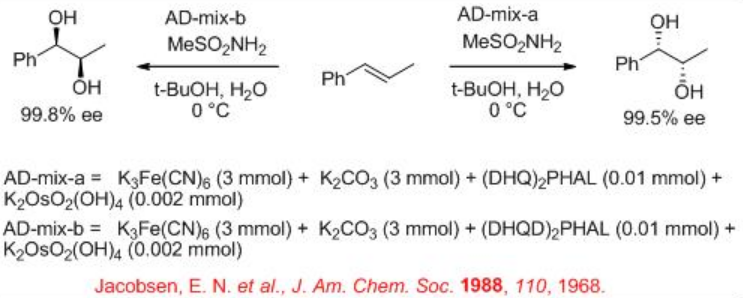
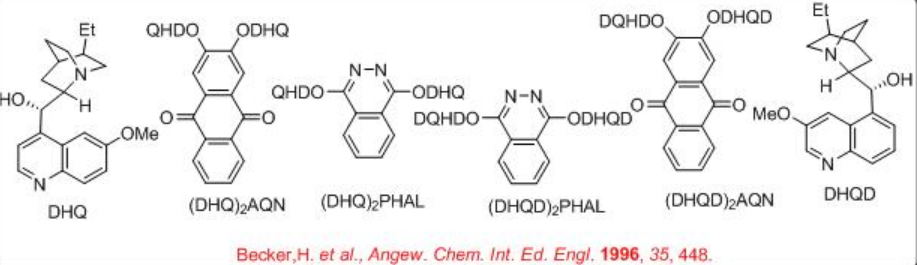
K3Fe(CN)6 is used as a oxidant to reoxidize the Os(VI) after each catalytic cycle. Since OsO4 is volatile and toxic, the osmium is usually added as K2OsO2 (OH)4, which forms OsO4 in the reaction mixture. K2CO3 and methanesulfonamide (MeSO2 NH2) are used as additive to enhance the rate of the reaction. Scheme 2 summarizes some of the successful cinchona alkaloid based ligands for the asymmetric dihydroxylation reactions. The approach of hydroxyl group is directed to either the top face or the bottom face of the alkene which depends on the nature of the ligands, DHQD or DHQ, are used.
In parallel to the above described catalytic processes, the use of optically active bidentate 1,2-diamine based ligand L has been demonstrated in place of alkaloid as a chiral source for the asymmetric dihydroxylation of alkenes using OsO4 (Scheme \(\PageIndex{3}\)). The reactions of a series of alkenes can be accomplished with good to excellent yield and enantioselectivity.
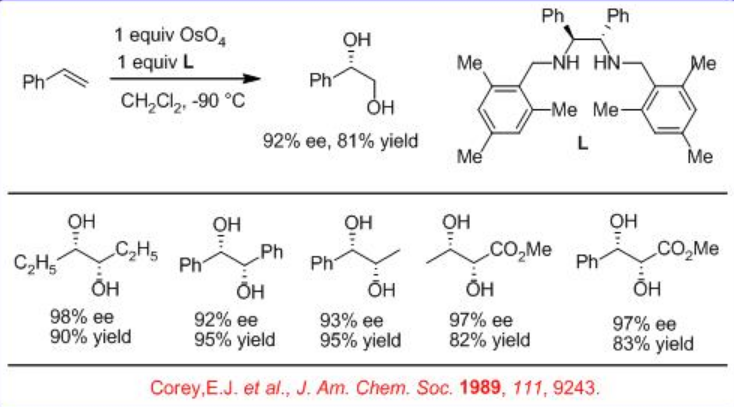
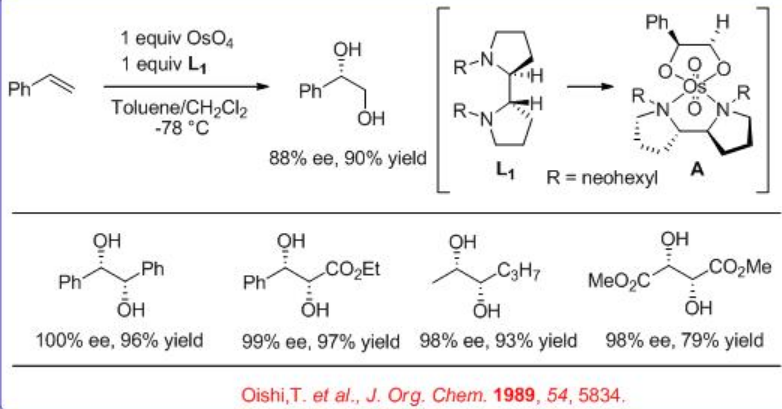
In addition, the bidentate ligand L 1 is found to effective for the OsO4 -mediated dihydroxylation of trans -disubstituted and monosubstituted alkenes (Scheme\(\PageIndex{4}\)). The reaction is believed to involve intermediate A and the products are obtained with high yield and enantioselectivity.
5.6.1.1 Synthesis of Biologically Important Molecules
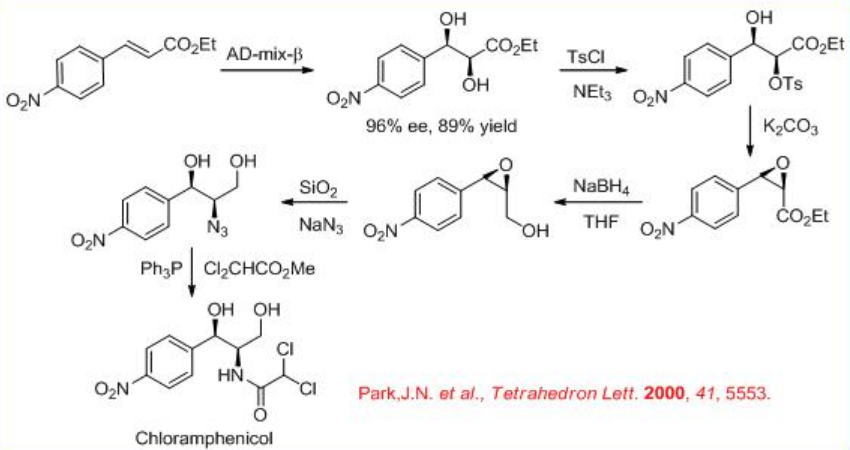
The Os-catalyzed enantioselective dihydroxylation is used as a key step in the highly expeditious synthesis of the antibacterial agent (–)-chloramphenicol (Scheme \(\PageIndex{5}\)).
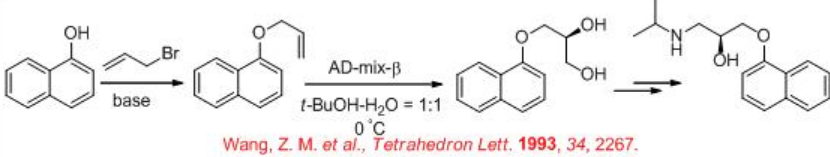
The synthesis of the β -receptor-blocking drug ( S ) - propranolol has been demonstrated employing osmium-catalyzed dihydroxylation as a key step (Scheme \(\PageIndex{6}\)). Reaction of α - naphthol with allylic bromide gives allyl naphthyl ether that could be dihydroxylated using AD-mix - β with 91% ee. The diol derivative could be converted into ( S ) - propranolol by classical methods.
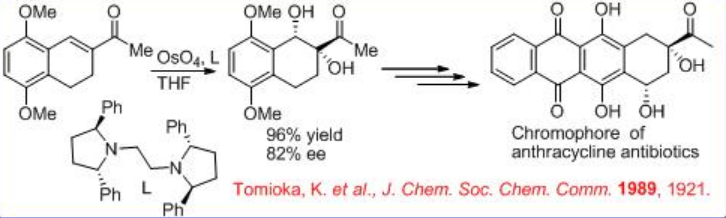
The synthesis of chromophore of anthracycline antibiotic uses chiral osmium complex bearing chiral diamine L for asymmetric dihydroxylation with good enantioselectivity (Scheme \(\PageIndex{7}\)). The resultant 1,2-diol could be subsequently converted into the desired chromophore of anthracycline antibiotic in good yield.
5.6.2 Asymmetric Aminohydroxylation
The chiral β-amino alcohol structural unit is a key motif in many biologically important molecules. It is difficult to imagine a more efficient means of creating this functionality than by the direct addition of the two heteroatom substituents to an alkene, especially if this transformation could be achieved in regioselective and enantioselective fashion. In parallel to allylic epoxidation and dihydroxylation of alkenes; Sharpless group has developed asymmetric aminohydroxylation of alkenes using osmium based catalysis.
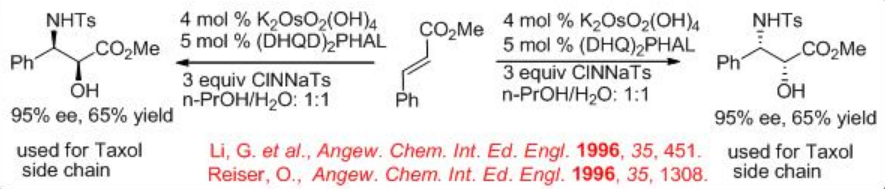
Synthesis of chiral α -sulfonamido hydroxy compounds can be obtained when the alkene substrates are subjected to the aminohydroxylation reaction using chloramine-T (TsNClNa) as the nitrogen source and H2O as the oxygen source. The reaction is found to be successful in the presence of osmium complex bearing (DHQ)2 PHAL or (DHQD)2 PHAL. The α -sulfonamido hydroxy compounds can be isolated with high yield and enantiomeric purity. Better results are obtained with chloramine-T (oxidant) salts bearing smaller organic substituents on the sulfur. This reagent could be prepared separately and added to the reaction mixture as the stable anhydrous salt or it can be generated in situ (Scheme \(\PageIndex{8}\)). The methyl (E )-cinnamate can be successfully converted into α-hydroxy- β-amino product with high enantioselectivity. The resultant product is used to construct the taxol side chain, and this process establishes the shortest and the most efficient route to the side chain of this pharmaceutically important agent.
The key issue is the regioselectivity of the reaction. Replacement of sulfonamide in chloramine-T with alkyl carbamates like BnO2CNH2 , EtO2CNH2, and t-BuO2CNH2 or amides greatly improves the reaction scope of the substrate and selectivity up to 99% ee and 80% yield. Also carbamate product could be easily converted into free amino alcohol. t -Butyl carbamate is superior to ethyl carbamate in terms of yield, enantioselectivity, and ease of removal of the N -protecting group.
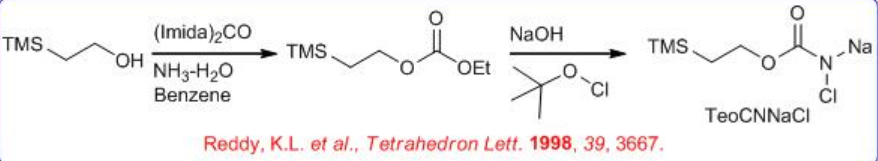
Nitrogen source 2-trimethylsilylethyl N -chloro- N -sodiocarbamate (TeoCNClNa) could be synthesized by reacting NaOH and t -BuOCl with 2-(trimethylsilyl)ethyl carbamate, which can be prepared by successively adding carbonyl diimidazole and ammonia to 2-trimethylsilylethanol in benzene (Scheme \(\PageIndex{9}\)-\(\PageIndex{10}\)). The TeoC group can be cleaved by fluoride under very mild conditions, yielding the free amino alcohol with high enantiomeric purity.

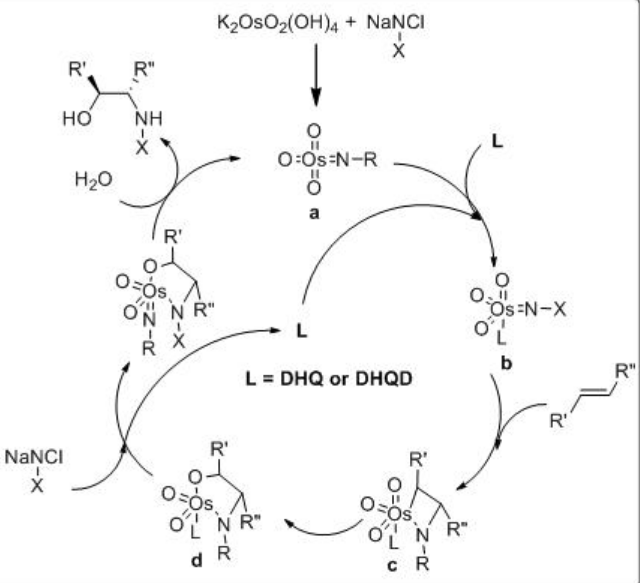
The mechanism of the reaction is shown in Scheme \(\PageIndex{11}\). The Os(VI) azaglycolate is reoxidized by the N -chloroamide substrate and releases the target product after hydrolysis. The reoxidized metallacycle undergoes a second cycloaddition leading to an Os(VI) bis(azaglycolate). Conducting the reaction in an aqueous medium under more dilute conditions favors the hydrolysis.
5.6.3 Asymmetric Aziridination
Aziridines are versatile building blocks in organic synthesis. Considerable progress has been made in the area of asymmetric aziridination employing copper based systems. Mn(porphyrin) and Mn-salen complexes have been shown as effective catalysts for this reaction. The reactions proceed via active nitrenoid species and most of the methods use a hypervalent iodine reagent such as PhI=NTs as nitrenoid source. The deprotection of N-sulfonyl groups require harsh reaction conditions, development of new methods has thus been focused without protecting group or with a readily removable group. In this context, the use of azide compounds as nitrogen source has been recently demonstrated.
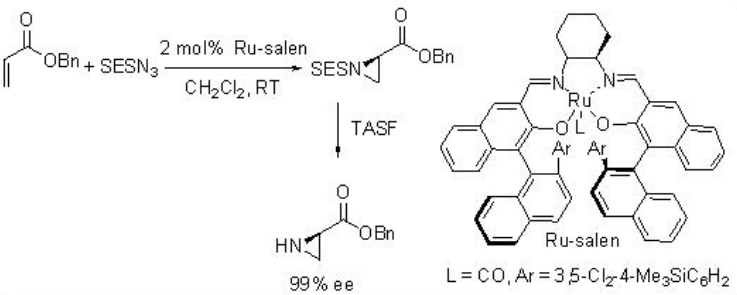
Ru-salen is found to be effective catalyst for the aziridination of alkenes with TsN3 at room temperature with excellent enantioselectivity (Scheme \(\PageIndex{12}\)). p -Nitro and o -nitrobenzenesulfonyl azide and 2-(trimethylsilyl)ethanesulfonyl azide (SESN3) are also effective for this reaction affording the aziridine with high enantioselectivity. Furthermore, less nucleophilic α, β -unsaturated esters proceed aziridination with high enantioselectivity.
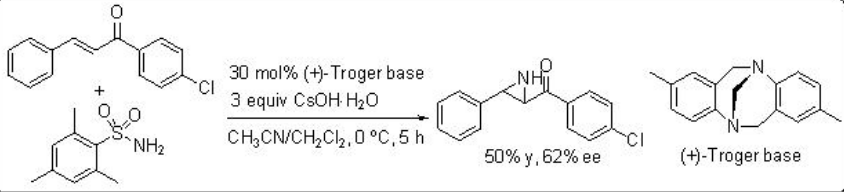
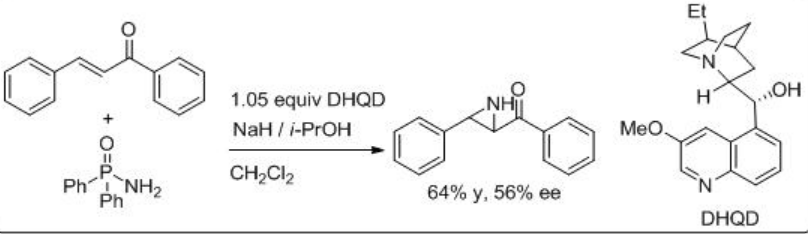
An aminimide that is generated by deprotonation of the corresponding aminimine undergoes aziridination of chalcone via conjugate addition and ring closure by N-N bond cleavage. For example, O-mesitylenesulfonylhydroxylamine proceeds reaction in the presence of (+)-Troger base and CsOH∙.H2O with moderate enantioselectivity (Scheme \(\PageIndex{13}\)). Soon after the use of quiniclidine for the reaction of O -(diphenylphosphinyl)hydroxylamine with chalcone is shown with 56% ee (Scheme \(\PageIndex{14}\)).