3.2: Reactions With Metal Nitrenoid and Direct C-H Oxidation
- Page ID
- 168781

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\dsum}{\displaystyle\sum\limits} \)
\( \newcommand{\dint}{\displaystyle\int\limits} \)
\( \newcommand{\dlim}{\displaystyle\lim\limits} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\(\newcommand{\longvect}{\overrightarrow}\)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)3.2.1 Reactions with Metal Nitrenoid
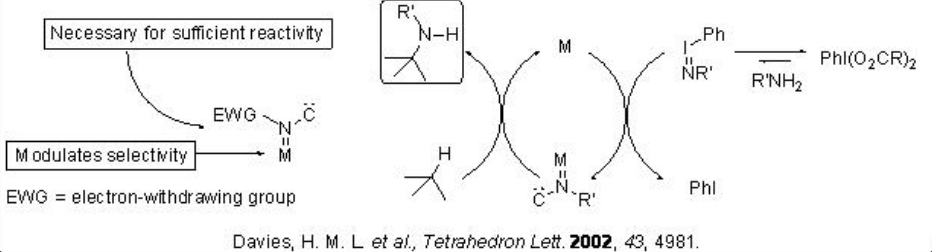
The amine functional group is an important component of many biologically active compounds. The nitrene insertion into C-H bonds provides powerful tool for the direct introduction of C-N bond from C-H bonds. The mechanism of metal nitrenoid formation is believed to take place via an in situ-formed iodonium ylide that produces the reactive metal nitrenoid intermediate in the presence of suitable metal (Scheme \(\PageIndex{1}\)). The major intrinsic factors controls the selectivity are (i) the catalyst and (ii) the electron withdrawing group.
3.2.1.1 Intramolecular Reactions
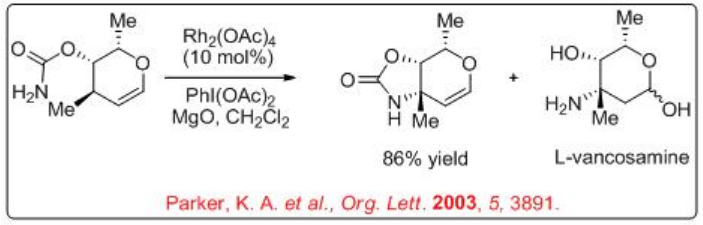
3-Amino glycol derivatives serve as intermediates for the synthesis of 2-oxygenated sugars, 2-deoxysugars and antibiotics. Dirhodium-catalyzed nitrene transfer has been utilized as a key step in the synthesis of carbamate-protected 3-aminoglycols. For example, Scheme \(\PageIndex{2}\) illustrates the selective transformation of carbamate into oxazolidinone via nitrene insertion with 86% yield. The resulting oxazolidinone can be converted into L-vancosamine. This method has been employed for the synthesis of protected glycols of L-daunosamine, D-saccharosamine and L-ristosamine.
Chiral Ru(II) porphyrin complex catalyzes the C-H amination of prochiral sulfonamides with good enantioselectivity (Scheme \(\PageIndex{3}\)). This procedure can be used for the synthesis of both five and six membered cyclic sulfamidates.
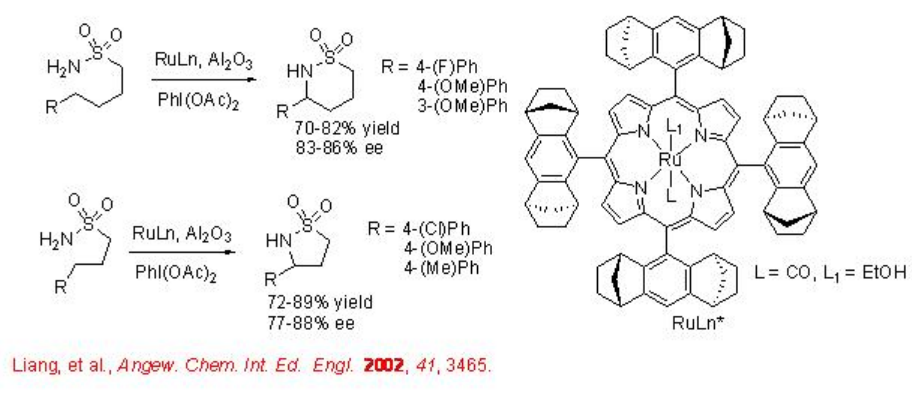
Chiral dirhodium has been shown effective catalyst for the cyclization of sulfonamides (Scheme \(\PageIndex{4}\)). This procedure is an example for the highly enantioselective amination process catalyzing the reactions of heteroaromatic substituents with up to 99% ee.
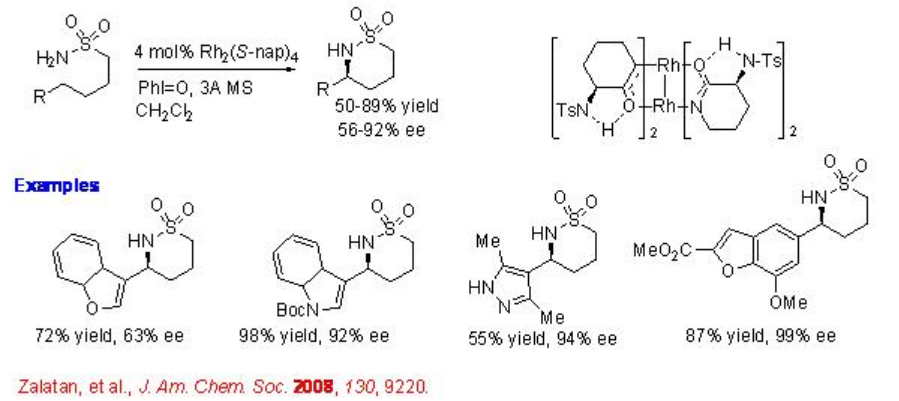
3.2.1.2 Intermolecular Reaction
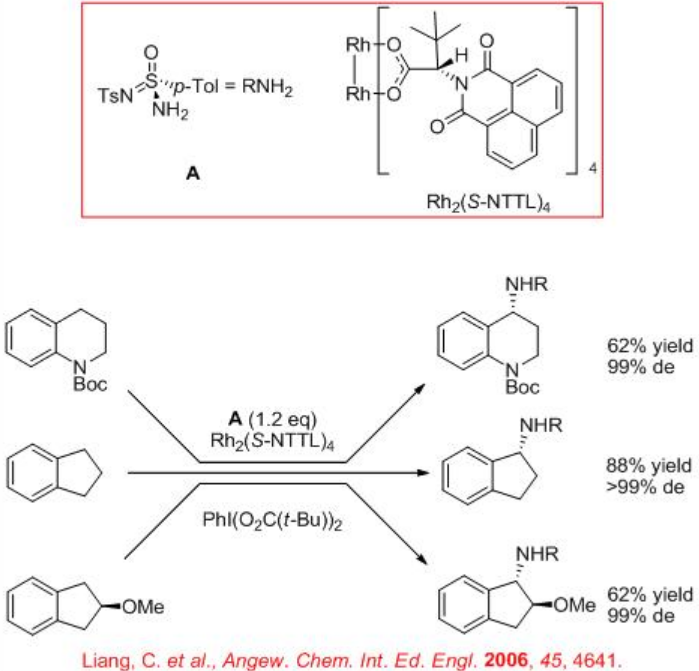
Intermolecular amination of benzylic C-H bonds can be accomplished using the chiral tosylsulfonylimidamide as a nitrene precursor in the presence of chiral dirhodium carboxylate Rh2(S-NTTL)4 with excellent diastereoselectivity (Scheme \(\PageIndex{5}\)).
3.2.2 C-H Activation via Direct C-H Oxidation
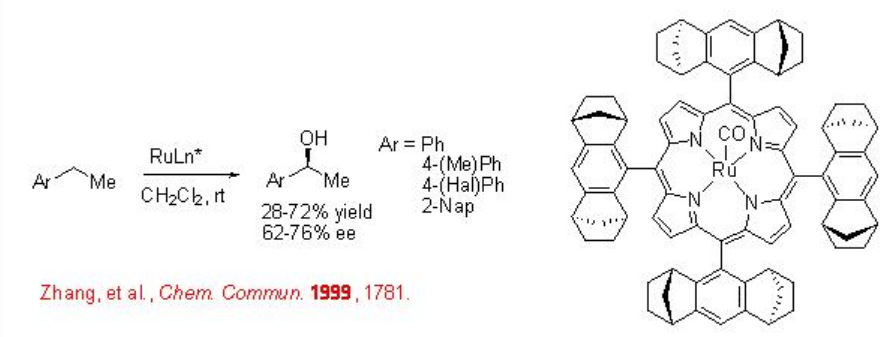
Chiral Ru-porphyrin complex has been shown to catalyze benzylic C-H hydroxylation with moderate enantioselectivity (Scheme \(\PageIndex{6}\)).
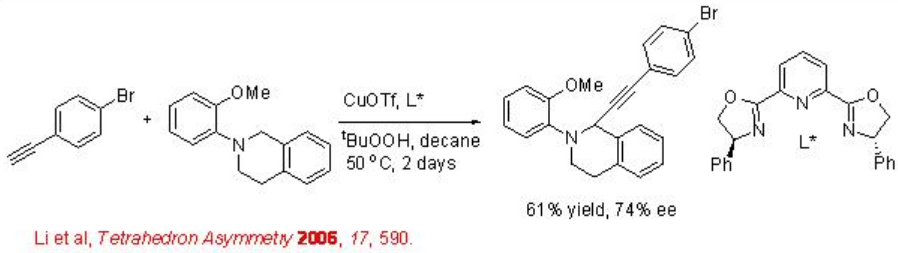
C-H functionalization via insertion of a reactive metal complex is one of the emerging areas for the development of practical C-H activation. The synthesis of alkynyl tetrahydroisoquinoline has been shown by double C-H activation in the presence of copper-pyBox at moderate temperature (Scheme \(\PageIndex{7}\)). The reaction of series of alkynes and aryl substituents is demonstrated. However, the presence of ortho methoxy substituent is essential for the success of the reaction.
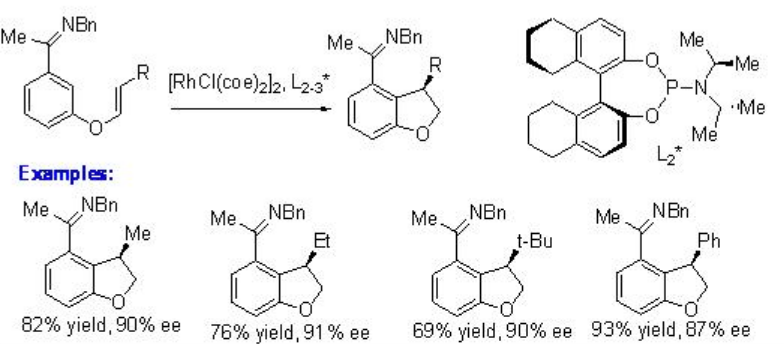
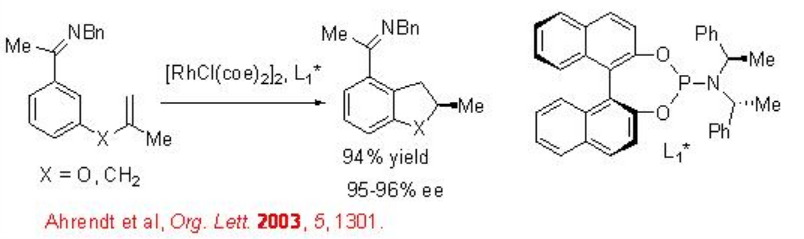
Intramolecular alkylation of ketimines has been shown using chiral rhodium complex bearing chiral phosphoramidite L1* (Scheme \(\PageIndex{8}\)). The observed results suggest that the reaction involves substrate directed oxidative addition of rhodium into the arene C-H bond. This approach provides a new cyclization strategy for the construction of five and six membered cyclic system.
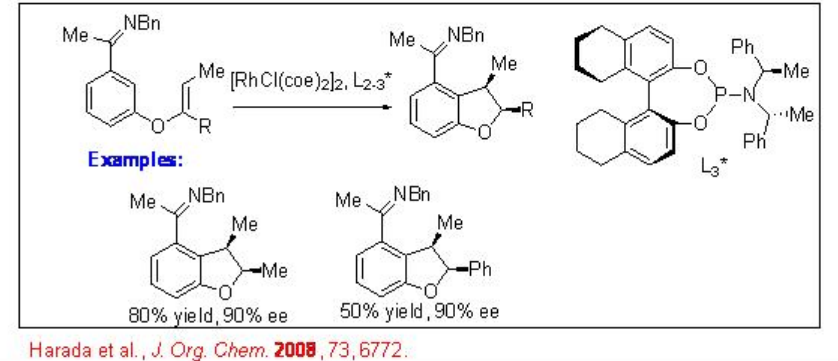
The scope of the above procedure has been expanded for the reactions of 1,2-disbstituted and 1,1,2-trisubstituted alkenes to give chiral indane, dihydrobenzofuran and dihydropyrroloindole with high enantioselectivity (Scheme \(\PageIndex{9}\)). The formation of syn -products is observed regardless the configuration of the starting alkenes. Thus, a mixture of E and Z alkenes may be used as starting material.