
13.1: Introduction to Organometallic Chemistry
- Page ID
- 385565

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)What is Organometallic Chemistry?
This chapter will introduce a subfield of inorganic coordination chemistry; organometallic chemistry. Let’s begin with a few simple questions: what is organometallic chemistry? What, after studying organometallic chemistry, will we know about the world that we didn’t know before? Why is the subject worth studying? And what kinds of problems is the subject meant to address? The purpose of this introduction is twofold: (1) to help motivate us as we move forward (that is, to remind us that there is a point to all this!); and (2) to illustrate the kinds of problems we’ll be able to address using concepts from the field. You might be surprised by the spine-chilling power you feel after learning about the behavior of organometallic compounds and reactions!
Put most bluntly, organometallic chemistry is the study of compounds containing, and reactions involving, metal-carbon bonds. The metal-carbon bond may be transient or temporary, but if one exists during a reaction or in a compound of interest, we’re squarely in the domain of organometallic chemistry. Despite the denotational importance of the M-C bond, bonds between metals and the other common elements of organic chemistry also appear in organometallic chemistry: metal-nitrogen, metal-oxygen, metal-halogen, and even metal-hydrogen bonds all play a role. Metals cover a vast swath of the periodic table and include the alkali metals (group 1), alkali earth metals (group 2), transition metals (groups 3-12), the main group metals (groups 13-15, “under the stairs”), and the lanthanides and actinides. We will focus most prominently on the behavior of the transition metals, so called because they cover the transition between the electropositive group 2 elements and the more electron-rich main group elements.
Why is organometallic chemistry worth studying? Well, for me, it mostly comes down to synthetic flexibility. There’s a reason the “organo” comes first in “organometallic chemistry”—our goal is usually the creation of new bonds in organic compounds. The metals tend to just be along for the ride (although their influence, obviously, is essential). And the fact is that you can do things with organometallic chemistry that you cannot do using straight-up organic chemistry. Case in point:
The venerable Suzuki reaction...unthinkable without palladium!
The establishment of the bond between the phenyl rings through a means other than dumb luck seems unthinkable to the organic chemist, but it’s natural for the palladium-equipped metal-organicker. Bromobenzene looks like a potential electrophile at the bromine-bearing carbon, and if you’re familiar with hydroboration you might see phenylboronic acid as a potential nucleophile at the boron-bearing carbon. Catalytic palladium makes it all happen! Organometallic chemistry is full of these mind-bending transformations, and can expand the synthetic toolbox of the organic chemist considerably.
To throw another motive into the mix for the non-specialist (or the synthesis-spurning chemist), organometallic chemistry is full of intriguing stories of scientific inquiry and discovery. Exploring how researchers take a new organometallic reaction from “ooh pretty” to strong predictive power is instructive for anyone interested in “how science works,” in a practical sense. We’ll examine a number of classical experiments in organometallic chemistry, both for their value to the field and their contributions to the general nature of scientific inquiry.
What kinds of problems should we be able to address as we move forward? Here’s a bulleted list of the most commonly encountered types of problems in an organometallic chemistry course:
- Describe the structure of an organometallic complex…
- Predict the product of the given reaction conditions…
- Draw a reasonable mechanism based on evidence…
- Devise a synthetic route to synthesize a target organometallic compound…
- Explain the observation(s)…
- Predict the results of a series of experiments…
The first four are pretty standard organic-esque problems, but it’s the last two, more general classes that really make organometallic chemistry compelling. Just imagine putting yourself in the shoes of the pioneers and making the same predictions they did!
There you have it, a short introduction to organometallic chemistry and why it’s worth studying. The rest of this chapter will describe what organometallic chemistry really is…it will be helpful to keep these motives in mind as you study. Keep a thirst for predictive power, and it’s hard to go wrong with organometallic chemistry!
Resources for learning organometallic chemistry
For the penny-pinching student or layman, there are several good resources for organometallic chemistry on the Web. Nothing as exhaustive as Reusch’s Virtual Textbook of Organic Chemistry exists for organometallic chemistry, but the base of resources available on the Web is growing. Rob Toreki’s Organometallic HyperTextBook could use a CSS refresh, but contains some nice introductions to different organometallic concepts and reactions. Try the electron-counting quiz!
VIPER is a collection of electronic resources for teaching and learning inorganic chemistry, and includes a nice section on organometallic chemistry featuring laboratory assignments, lecture notes, and classroom activities. Awesome public lecture notes are available from Budzelaar at the University of Manitoba and Shaughnessy at Alabama (Roll Tide?). For practice problems, check out Fu’s OpenCourseWare material from MIT and Shaughnessy’s problem sets.
Historical Background and Introduction to Metallocenes
Organometallic Complex: A complex with bonding interactions between a metal atom and one or more carbon atoms of an organic group or molecule.
An organometallic complex is defined as a complex with bonding interactions between one or more carbon atoms of an organic group or molecule and at least one metal atom. It is important to understand that just the presence of an organic ligand is not sufficient to define an organometallic compound. If there are organic ligands present, but no metal-carbon bonds, the complex is said to be "metal-organic". There must be interactions between a carbon and a metal for the complex to be considered organometallic.
Which of the molecules below is an organometallic complex?
\[\ce{[Sn(Me)4]} \quad \quad \ce{Sn(OMe)4} \ce{cis-[PtCl2(NH3)2]}\]
Solution
The tetramethyl tin (\(\ce{Sn(Me)4}\)) has metal-carbon bonds, and thus it is an organometallic complex.
Tetramethoxy tin (\(\ce{Sn(OMe)4}\)) has metal-oxygen bonds. It lacks metal-carbon bonds, but it does contain organic ligands; thus it is a "metal-organic" complex rather than organometallic.
Cisplatinum (\(\ce{cis-[PtCl2(NH3)2]}\)) contains no carbon and thus it is neither organometallic or metal-organic; it is just a coordination compound.
Industrial Importance of Organometallic Compounds
Organometallic compounds are a very important class of compounds in industry. For example, tens of thousand of tons of aluminum and tin alkyl compounds are produced each year for industrial purposes. The use of organometallic compounds as catalysts to produce other compounds is even more important. Organometallic catalysts are used for a range of industrial syntheses from manufacture of commodity polymers like polypropylene and polyethylene to production of simple organic molecules like acetaldehyde and acetic acid. These compounds are produced at a scale in the order of millions of tons per year.
History of Organometallic Chemistry
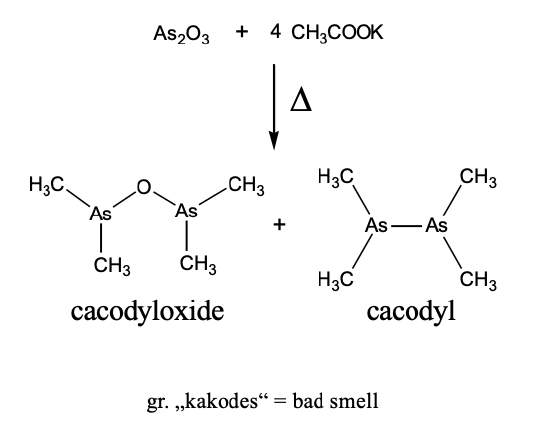
Let us take a historical approach to organometallic chemistry and see how the field has evolved. Arguably, the first organometallic compound was kakodyl oxide, an organo-arsenic compound (Figure \(\PageIndex{2}\)). It was accidentally produced by the French chemist Louis Cadet in 1760 when he was working on inks. He heated arsenic oxide and potassium acetate and obtained a red-brown oily liquid, known as Cadet’s fuming liquid. It consists mostly of cacodyl and cacodyl oxide (Figure \(\PageIndex{2}\)). In cacodyl, there is an As-As bond and two methyl groups attached to each As atom. In the other product, cacodyl oxide, there is an O atom bridging the two As atoms, and each As is bound to two methyl groups. The names of these two compounds come from the greek word “kakodes”, meaning "bad smell". Indeed, they have a very intense, garlic-like smell. The reaction can be used to identify arsenic in samples. For instance, if you suspected your food was poisoned with arsenic, you could heat a sample together with potassium acetate. If a bad, garlic-like smell evolved, this would indicate that there is arsenic in your sample.
Another important milestone in coordination chemistry was the discovery of Zeise’s salt by the Danish chemist William Zeise in 1827 (Figure \(\PageIndex{3}\)). Zeise’s salt was the first olefin complex in which an olefin was bound side-on to a metal using its π-electrons for σ-bonding. Zeise observed that when sodium hexachloroplatinate (2-) was heated in ethanol, a compound with the composition Na[PtCl3(C2H4)] could be isolated. The chemical composition of the compound suggested that there was a C2H4 organic fragment bonded to the platinum, but it was not clear how.
This question was only answered more than 100 years later in 1969 with the crystal structure analysis of Zeise’s salt. The crystal structure revealed that an ethylene molecule was bound side-on to the platinum (Figure \(\PageIndex{3}\)). The two carbon atoms had about the same distance to the Pt arguing that they were equally strongly involved in the bonding with Pt. The first C had a distance of 216 pm while the second carbon had a distance of 215 pm. The ethylene molecule was oriented perpendicular to the plane made of the three chloro ligands and the platinum atom. The Cl-Pt-Cl bond angles were near 90°. Overall, the structure could be described as a structure derived from a square planar structure with the ethylene as the fourth ligand that would stand perpendicular to the plane in order to minimize steric repulsion with the chloro ligands.
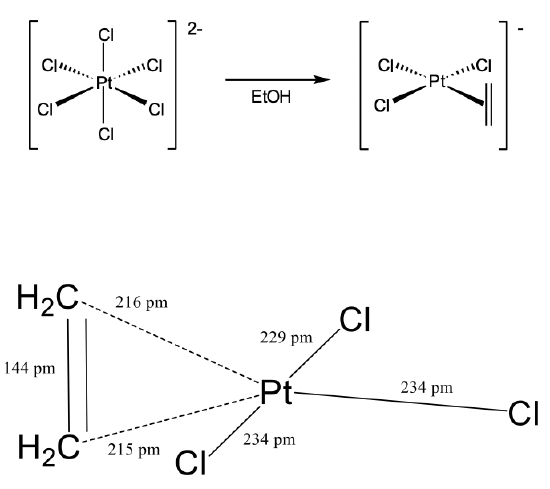
An important question was how to describe the bonding in the compound. It was noteworthy that the C-C bond length in Zeise’s salt was significantly longer (144 pm) than that in a free ethylene molecule (134 pm). Based on the bond-strength bond-length concept this argued that the bond was weaker than a regular C=C double bond, and that the bond order was smaller than 2. What could explain this lower bond order and the side-on coordination of the ethylene in Zeise’s salt?
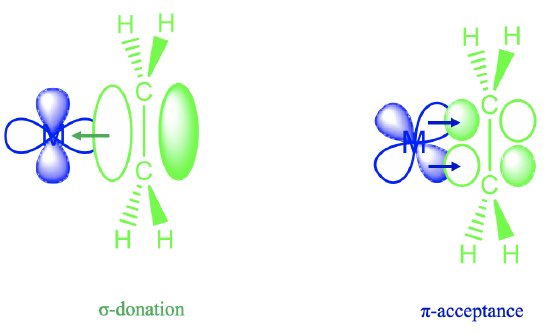
The answer is that the ethylene molecule uses its π-electrons for σ-bonding with a metal d-orbital (Figure \(\PageIndex{4}\)). You can see above that the lobe of a dx2-y2 orbital has the correct orientation to overlap with the bonding π-orbitals of the ethylene ligand in a σ-fashion. Through this interaction electron density gets donated from the bonding ligand π-orbital into the metal-d-orbital. This results in a lower electron density for π-bonding within the ligand, and thus the bond order is decreased. In addition, there is another effect that lowers the bond order. A metal d-orbital can interact with the π*-orbitals in π-fashion. In this process, electron density can be donated from the metal d-orbital into the empty π-orbitals of the ligand. The increased electron density in the π-orbitals further decreases the bond order.
In 1890, a further milestone in organometallic chemistry was reached with the synthesis of the first carbonyl complex, the nickel tetracarbonyl. Nickel tetracarbonyl is a colorless liquid that boils at 43°C. The compound was prepared by the German chemist Ludwig Mond. Nickel tetracarbonyl forms spontaneously from nickel metal and carbon monoxide at room temperature. The reaction is the basis of the Mond-process that is used up to date in order to purify nickel (Figure \(\PageIndex{5}\)). Carbon monoxide gas can be streamed over impure Ni metal at temperatures of 50-60°C to form Ni(CO)4 in gaseous form. Impurities are left behind in solid form. Then, the nickel tetracarbonyl is thermally decomposed at ca. 220° to form nickel and carbon monoxide. The process can be varied to produce nickel in powder form, as spheres (Figure \(\PageIndex{5}\)), and as coatings. The Mond process is still used despite the very high toxicity of nickel tetracarbonyl.
\[\begin{align*}
\ce{Ni_{(s, ~ impure)} + 4 CO_{(g)}} &\ce{ -> Ni(CO)4_{(g)}} & &
50-60^{\circ} \text{C (also spontaneous at room temp)}
\\[5pt]
\ce{Ni(CO)4_{(g)}} &\ce{ -> Ni_{(s, ~ purified)} + 4 CO} & &220-250 ^{\circ} \text{C (Ni is purified)}
\end{align*}\]

In 1900 the first Grignard reagents were discovered. Victor Grignard (Figure \(\PageIndex{9}\)) was an enthusiastic young French chemist who discovered how to make organomagnesium halides (RMgX) while working for his Ph.D.. His supervisor, Sabatier, had been trying this chemistry for some time, but Victor was the genius who solved the problem. Organomagnesium halides form from magnesium and organic halides in ethers. This discovery in 1900 changed the course of organic chemistry and won Grignard and Sabatier the Nobel Prize in 1912.
\[\text{The Grignard Reaction:} \quad \ce{Mg_{(s)}} + R-X ->[ether] R-Mg-X}\]
In 1917, the first lithium alkyls were prepared by Wilhelm Schlenk. Like Grignard reagents, lithium alkyls are very valuable reactants in synthetic organic chemistry. Lithium alkyls are very air-sensitive compounds, and some even self-ignite in air (for example tert-butyl lithium is explosive when exposed to air). Therefore, they need to be handled carefully under inert gas. So that the reactions could be carried out under air0free conditions, Wilhelm Schlenk made an important invention: The Schlenk-lines (Figure \(\PageIndex{6}\)). The Schlenk lines allows a chemist to remove all gas (air) from a reaction flasks and back-fill the flask with an inert gas. A Schlenk line has a dual manifold with several ports. One manifold is connected to a source of purified inert gas, while the other is connected to a vacuum pump. The inert-gas line is vented through an oil bubbler, while solvent vapors and gaseous reaction products are prevented from contaminating the vacuum pump by a liquid nitrogen or dry ice/acetone cold trap. Special stopcocks or Teflon taps allow vacuum or inert gas to be selected without the need for placing the sample on a separate line.

Discovery of Metallocenes
The discoveries discussed above were important milestones in the development of organometallic chemistry. But the field expanded more rapidly with the discovery of the first metallocene: ferrocene. Like often in science, ferrocene was discovered through an accident. In 1951 Peter Pauson and Thomas Kealy attempted to synthesize the organic compound fulvalene through oxidative coupling of cyclopentadienyl magnesium chloride with iron (III) chloride. However, instead of obtaining fulvalene they observed the formation hydrofulvalene together with an orange powder with “remarkable stability”. Analysis of the powder showed that the chemical compound contained two cyclopentadienyl rings and one iron atom. According to the knowledge on bonding in organometallic compounds that was known at the time Kealy and Pauson suggested a structure for the molecule in which two cyclopentadienyl group were bound to a single Fe atom via two Fe-C bonds (Figure \(\PageIndex{7}\)).
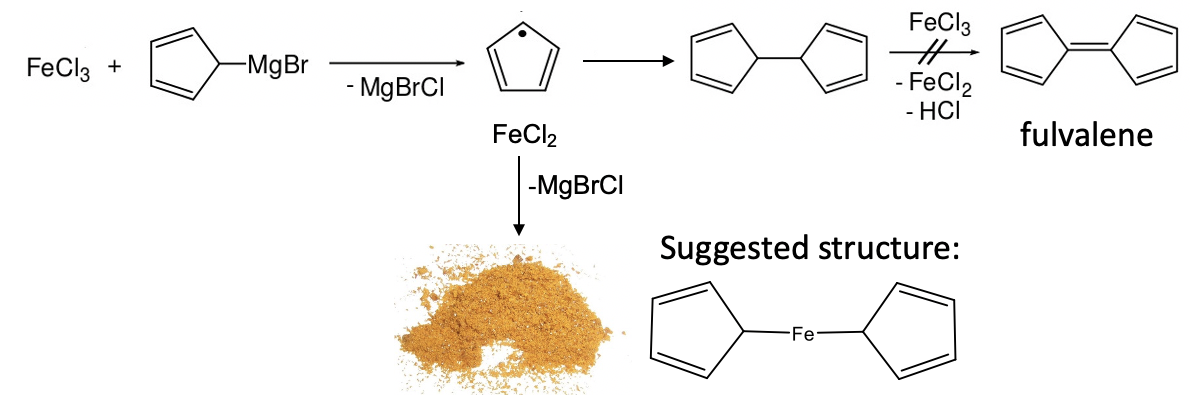
However, the remarkable stability of the compound was in contradiction to the proposed structure. The structure would be a 10 electron complex which would be highly coordinatively unsaturated. This would be inconsistent with the observed stability. We can briefly practice our electron counting skills again in order to prove that there are only 10 electrons (Figure \(\PageIndex{8}\)). (Electron counting will be discussed in more depth later in this chapter)
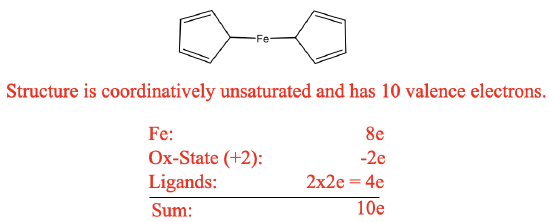
The compound was stable in air, and could be sublimed without decomposition. Further, the double-bonds in the cyclopentadienyl rings resisted hydrogenation. In addition, the structure was inconsistent with spectroscopic observations. Only one C-H stretch vibration was observed in the IR and only one signal was observed in the 1H NMR. Kealy and Pauson’s structure would have been consistent with three C-H stretch vibrations and three NMR resonances. For these reasons, Kealy’s and Pausons structure was soon questioned. Ernst Otto Fischer at University of Munich and Sir Geoffrey Wilkinson at Harvard University (Figure \(\PageIndex{9}\)) suggested an alternative structure that was very different from all known organometallic structures. The bonding in this structure was completely different from all bonding concepts considered thus far for organometallics. What structure did they propose? They proposed a sandwich structure in which the two aromatic cyclopentadienyl anions would sandwich an Fe2+ ion. What would be the bonding in this structure? The cylopentadienyl anion would donate its six π-electrons into the valence orbitals of the metal. Thus, all five carbon atoms would be equally involved in the bonding, the ligand would act as a ƞ5-ligand. Because of the donation of π-electrons in the cycopentadiene unit, the structure was called “ferrocene”. The ferrocene structure would explain the stability and low reactivity of the compound.
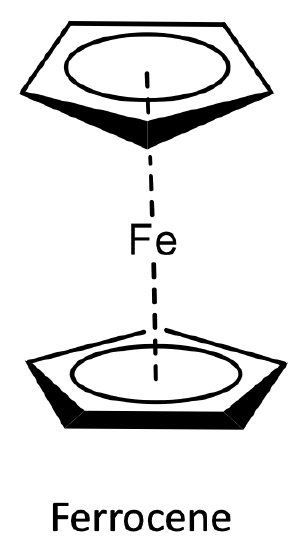
Electron-counting gives an 18 electron complex (Figure \(\PageIndex{10}\)). Let us verify this using the oxidation state method. Fe would contribute eight electrons in the neutral state. The ligands would be considered as cyclopendienyl anions with a 1- charge. This would give an oxidation state of +2 for Fe reducing the number of electrons from eight to six. The two ligands would contribute six electrons each giving twelve electrons. 12+6=18. Hence, the structure fulfills the 18 electron rule. The structure is also consistent with the spectroscopic observation of one NMR resonance and one C-H stretch vibration. All these were excellent arguments to support the ferrocene sandwich structure, however they were not an absolute proof. The proof finally came with the X-ray crystal structure determination which unambiguously confirmed the sandwich structure.
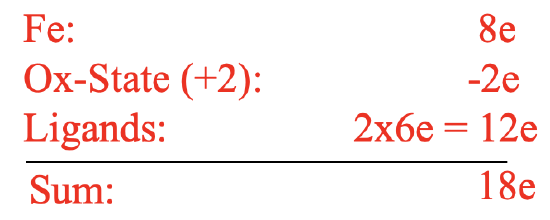
Dr. Kai Landskron (Lehigh University). If you like this textbook, please consider to make a donation to support the author's research at Lehigh University: Click Here to Donate.