
5.6: Crystal Field Stabilization Energy, Pairing, and Hund's Rule
- Page ID
- 183322

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)The splitting of the d-orbitals into different energy levels in transition metal complexes has important consequences for their stability, reactivity, and magnetic properties. Let us first consider the simple case of the octahedral complexes [M(H2O)6]3+, where M = Ti, V, Cr. Because the complexes are octahedral, they all have the same energy level diagram:
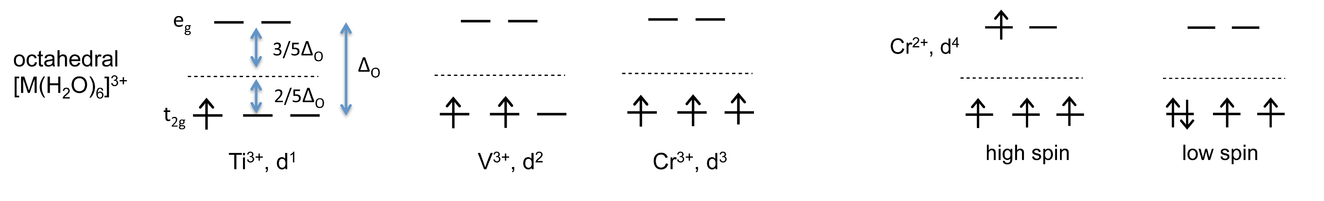
The Ti3+, V3+, and Cr3+ complexes have one, two and three d-electrons respectively, which fill the degenerate t2g orbitals singly. The spins align parallel according to Hund's rule, which states that the lowest energy state has the highest spin angular momentum.
For each of these complexes we can calculate a crystal field stabilization energy, CFSE, which is the energy difference between the complex in its ground state and in a hypothetical state in which all five d-orbitals are at the energy barycenter.
For Ti3+, there is one electron stabilized by 2/5 ΔO, so \(CFSE= -(1)(\frac{2}{5})(\Delta_{O}) = \frac{-2}{5} \Delta_{O}\)
Similarly, CFSE = -4/5 ΔO and -6/5 ΔO for V3+ and Cr3+, respectively.
For Cr2+ complexes, which have four d-electrons, the situation is more complicated. Now we can have a high spin configuration (t2g)3(eg)1, or a low spin configuration (t2g)4(eg)0 in which two of the electrons are paired. What are the energies of these two states?
High spin: \(CFSE= (-3)(\frac{2}{5})\Delta_{O} + (1)(\frac{3}{5})\Delta_{O} = -\frac{3}{5}\Delta_{O}\)
Low spin: \(CFSE= (-4)(\frac{2}{5})\Delta_{O} + P = -\frac{8}{5} \Delta_{O} + P\), where P is the pairing energy
Energy difference = -ΔO + P
The pairing energy P is the energy penalty for putting two electrons in the same orbital, resulting from the electrostatic repulsion between electrons. For 3d elements, a typical value of P is about 15,000 cm-1.
The important result here is that a complex will be low spin if ΔO > P, and high spin if ΔO < P.
Because ΔO depends on both the metals and the ligands, it determines the spin state of the complex.
Rules of thumb:
3d complexes are high spin with weak field ligands and low spin with strong field ligands.
High valent 3d complexes (e.g., Co3+ complexes) tend to be low spin (large ΔO)
4d and 5d complexes are always low spin (large ΔO)
Note that high and low spin states occur only for 3d metal complexes with between 4 and 7 d-electrons. Complexes with 1 to 3 d-electrons can accommodate all electrons in individual orbitals in the t2g set. Complexes with 8, 9, or 10 d-electrons will always have completely filled t2g orbitals and 2-4 electrons in the eg set.
|
d-orbital energy diagrams for high and low spin Co2+ complexes, d7 |
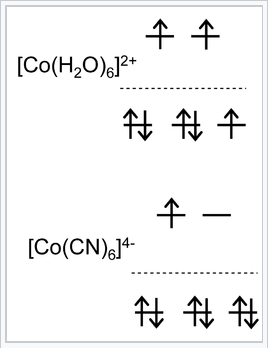
Examples of high and low spin complexes:
[Co(H2O)62+] contains a d7 metal ion with a weak field ligand. This complex is known to be high spin from magnetic susceptibility measurements, which detect three unpaired electrons per molecule. Its orbital occupancy is (t2g)5(eg)2.
We can calculate the CFSE as \(-(5)(\frac{2}{5})\Delta_{O} + (2)(\frac{3}{5})\Delta_{O} = -\frac{4}{5} \Delta_{O}\)
[Co(CN)64-] is also an octahedral d7 complex but it contains CN-, a strong field ligand. Its orbital occupancy is (t2g)6(eg)1 and it therefore has one unpaired electron.
In this case the CFSE is \(-(6)(\frac{2}{5})\Delta_{O} + (1)(\frac{3}{5})\Delta_{O} + P = -\frac{9}{5}\Delta_{O} + P.\)
Magnetism of transition metal complexes
Compounds with unpaired electrons have an inherent magnetic moment that arises from the electron spin. Such compounds interact strongly with applied magnetic fields. Their magnetic susceptibility provides a simple way to measure the number of unpaired electrons in a transition metal complex.
If a transition metal complex has no unpaired electrons, it is diamagnetic and is weakly repelled from the high field region of an inhomogeneous magnetic field. Complexes with unpaired electrons are typically paramagnetic. The spins in paramagnets align independently in an applied magnetic field but do not align spontaneously in the absence of a field. Such compounds are attracted to a magnet, i.e., they are drawn into the high field region of an inhomogeneous field. The attractive force, which can be measured with a Guoy balance or a SQUID magnetometer, is proportional to the magnetic susceptibility (χ) of the complex.
The effective magnetic moment of an ion (µeff), in the absence of spin-orbit coupling, is given by the sum of its spin and orbital moments:
\[\mathbf{\mu_{eff}= \mu_{spin} + \mu_{orbital} = \mu_{s} + \mu_{L}}\]
In octahedral 3d metal complexes, the orbital angular momentum is largely "quenched" by symmetry, so we can approximate:
\[\mathbf{\mu_{eff} \approx \mu_{s}}\]
We can calculate µs from the number of unpaired electrons (n) using:
\[\mu_{eff} = \sqrt{n(n+2)}\mu_{B}\]
Here µB is the Bohr magneton (= eh/4πme) = 9.3 x 10-24 J/T. This spin-only formula is a good approximation for first-row transition metal complexes, especially high spin complexes. The table below compares calculated and experimentally measured values of µeff for octahedral complexes with 1-5 unpaired electrons.
| Ion | Number of unpaired electrons |
Spin-only moment /μB |
observed moment /μB |
|---|---|---|---|
| Ti3+ | 1 | 1.73 | 1.73 |
| V4+ | 1 | 1.68–1.78 | |
| Cu2+ | 1 | 1.70–2.20 | |
| V3+ | 2 | 2.83 | 2.75–2.85 |
| Ni2+ | 2 | 2.8–3.5 | |
| V2+ | 3 | 3.87 | 3.80–3.90 |
| Cr3+ | 3 | 3.70–3.90 | |
| Co2+ | 3 | 4.3–5.0 | |
| Mn4+ | 3 | 3.80–4.0 | |
| Cr2+ | 4 | 4.90 | 4.75–4.90 |
| Fe2+ | 4 | 5.1–5.7 | |
| Mn2+ | 5 | 5.92 | 5.65–6.10 |
| Fe3+ | 5 | 5.7–6.0 |
The small deviations from the spin-only formula for these octahedral complexes can result from the neglect of orbital angular momentum or of spin-orbit coupling. Tetrahedral d3, d4, d8 and d9 complexes tend to show larger deviations from the spin-only formula than octahedral complexes of the same ion because quenching of the orbital contribution is less effective in the tetrahedral case.
Summary of rules for high and low spin complexes:
3d complexes: Can be high or low spin, depending on the ligand (d4, d5, d6, d7)
4d and 5d complexes: Always low spin, because ΔO is large
Maximum CFSE is for d3 and d8 cases (e.g., Cr3+, Ni2+) with weak field ligands (H2O, O2-, F-,...) and for d3-d6 with strong field ligands (Fe2+, Ru2+, Os2+, Co3+, Rh3+, Ir3+,...)
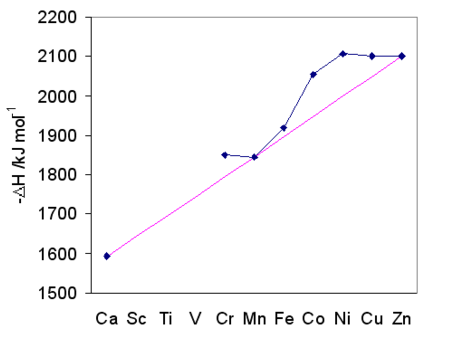
Irving-Williams series. For M2+ complexes, the stability of the complex follows the order Mg2+ < Mn2+ < Fe2+ < Co2+ < Ni2+ < Cu2+ > Zn2+. This trend represents increasing Lewis acidity as the ions become smaller (going left to right in the periodic table) as well as the trend in CFSE. This same trend is reflected in the hydration enthalpy of gas-phase M2+ ions, as illustrated in the graph at the right. Note that Ca2+, Mn2+, and Zn2+, which are d0, d5(high spin), and d10 aquo ions, respectively, all have zero CFSE and fall on the same line. Ions that deviate the most from the line such as Ni2+ (octahedral d8) have the highest CFSE.
Colors and spectra of transition metal complexes
Transition metal complexes often have beautiful colors because, as noted above, their d-d transition energies can be in the visible part of the spectrum. With octahedral complexes these colors are faint (the transitions are weak) because they violate the Laporte selection rule. According to this rule, g -> g and u -> u transitions are forbidden in centrosymmetric complexes. d-orbitals have g (gerade) symmetry, so d-d transitions are Laporte-forbidden. However octahedral complexes can absorb light when they momentarily distort away from centrosymmetry as the molecule vibrates. Spin flips are also forbidden in optical transitions by the spin selection rule, so the excited state will always have the same spin multiplicity as the ground state.
The spectra of even the simplest transition metal complexes are rather complicated because of the many possible ways in which the d-electrons can fill the t2g and eg orbitals. For example, if we consider a d2 complex such as V3+(aq), we know that the two electrons can reside in any of the five d-orbitals, and can either be spin-up or spin-down. There are actually 45 different such arrangements (called microstates) that do not violate the Pauli exclusion principle for a d2 complex. Usually we are concerned only with the six of lowest energy, in which both electrons occupy individual orbitals in the t2g set and all their spins are aligned either up or down.
|
From left: [V(H2O)6]2+ (lilac), [V(H2O)6]3+ (green), [VO(H2O)5]2+ (blue) and [VO(H2O)5]3+ (yellow). |

We can see how these microstates play a role in electronic spectra when we consider the d-d transitions of the [Cr(NH3)6]3+ ion. This ion is d3, so each of the three t2g orbitals contains one unpaired electron. We expect to see a transition when one of the three electrons in the t2g orbitals is excited to an empty eg orbital. Interestingly, we find not one but two transitions in the visible.
The reason that we see two transitions is that the electron can come from any one of the t2g orbitals and end up in either of the eg orbitals. Let us assume for the sake of argument that the electron is initially in the dxy orbital. It can be excited to either the dz2 or the dx2-y2 orbital:
\(d_{xy} \rightarrow d_{z^{2}}\) (higher energy)
\(d_{xy} \rightarrow d_{x2-y2}\) (lower energy)
The first transition is at higher energy (shorter wavelength) because in the excited state the configuration is (dyz1dxz1dz21). All three of the excited state orbitals have some z-component, so the d-electron density is "piled up" along the z-axis. The energy of this transition is thus increased by electron-electron repulsion. In the second case, the excited state configuration is (dyz1dxz1dx2-y21), and the d-electrons are more symmetrically distributed around the metal. This effect is responsible for a splitting of the d-d bands by about 8,000 cm-1. We can show that all other possible transitions are equivalent to one of these two by symmetry, and hence we see only two visible absorption bands for Cr3+ complexes.