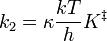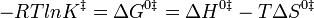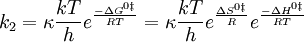5.1: Hydrogen, Oxygen, and Water
- Page ID
- 46891
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
Chemical phenomena must be treated as if
- Lothar Meyer (1868)
Introduction
The goal of every chemist, no matter what types of chemical compounds he or she works with, is to understand how and why chemicals react and change. Yet this is the most difficult task of all. It is not enough to know the structures of all the reactants and products, although such knowledge is a vital starting point. We must also know how the molecules approach one another and with what energies and with what orientations they interact. The concepts of energy and entropy are important in understanding chemical reactions. In this chapter we shall look at some of the problems that face us because we cannot examine individual molecular events. We shall examine two theories for predicting the rates of such simple reactions and compare their success or lack of it. We shall look at the two factors that often make reactions slow - energy and entropy - and see how catalysts can overcome these factors and accelerate chemical changes. Although we cannot present a complete theory of chemical reaction (no one can do this yet), we can outline the foundations on which this theory will someday be constructed.
What Happens When Molecules React?
Mechanisms of Reaction
Let’s suppose that we can watch what happens when two molecules react. We can take as an example the reaction of a molecule of thioacetamide, CH3—CS—NH2, with water to yield acetamide, CH3—CO—NH2, and H2S (Figure 22-1).
thioacetamide, with water to make acetamide, and H2S, (a) The thioacetamide molecule has S, C and N in one plane w around a central C. The orbitals of the double bond are distorted toward S to represent its greater elecironegativity. Orbitals that play no part in the reaction are not drawn.]]
state, with partial bonds from C to both S and O. The former 0—H bonding electron pairs are becoming lone pairs. (c) Products of the reacuon acetamide and H2S. The tetrahedral geometry of the transition state has reverted to trigonal planar geometry as the S atom leaves. The bond angle in H2S is 92° in contrast to the 105° in (a)]]In the original thioacetamide molecule, the central carbon atom is bound to carbon and to nitrogen by σ bonds, and by a σ and a π double bond to sulfur (Figure 22-1a). Since sulfur and nitrogen are more electronegative than carbon, electrons are more attracted to these atoms. Thus the electron pairs of the carbon-sulfur and carbon-nitrogen bonds their bonds are slightly displaced toward sulfur and nitrogen, causing these two atoms to bear a small negative charge and the central carbon to bear a small positive charge.
The most favorable direction of approach of a water molecule is perpendicularly from either side of the plane of the four heavy atoms. The most favorable orientation of the incoming water molecule is as shown in Figure 22-1a. Here a lone electron pair from the water is attracted to the positive charge on the central carbon. As the water molecule approaches this carbon atom, the lone-pair electrons are drawn to it and begin to form a partial bond. This partial bond formation has two effects: It weakens the bond between carbon and sulfur by repelling the electrons even more toward sulfur, and it simultaneously weakens the O—H bonds in water by pulling electrons from these bonds toward oxygen as the oxygen lone-pair electrons are attracted toward carbon. This intermediate state appears in Figure 22-1b. The central carbon atom now has two single bonds to carbon and nitrogen, and two partial bonds to sulfur and oxygen.
This intermediate state is not stable. If the water molecule falls away again and the situation reverts to that of Figure 22-1a (and there is no reason why this could not happen), then we see no net reaction. The water molecule rebounds from a collision with thioacetamide and goes its separate way. It also could happen that the sulfur atom falls away, as in Figure 22-1c. In this reaction, the two protons released by oxygen as it makes a double bond with carbon are attracted by the sulfur atom with four electron pairs, and a molecule of H2S results. The reaction
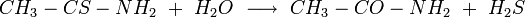
is complete. The reverse reaction also can occur; a molecule of H2S can collide with one of acetamide and produce water and thioacetamide. We would be less likely to see such an event if we could watch reactions at the molecular level, simply because there are very few H2S molecules in comparison to the number of water molecules.
Factors of Reactions
Geometry of approach is a major factor in reactions . If the water molecule approached in the plane of the thioacetamide molecule, it would find its entry blocked by sulfur lone electron pairs and hydrogen atoms (to a greater extent than is apparent in the skeletal drawings of Figure 22-1). Moreover, if the water molecule approached with a hydrogen atom, instead of a lone electron pair, pointed at the central carbon, it would not be as attracted to the thioacetamide molecule and would be more likely to rebound without reacting. If we could watch every collision, we might see that only 1 collision in 10, or in 100, had both molecules properly oriented for reaction.
A second factor is the energy of the two molecules. In the simplest theories this is expressed only as the relative speed of the two molecules upon collision. If the relative speed of the two molecules is small upon impact, the intermediate state will be more likely to revert to the starting molecules. A slowly moving water molecule can bounce harmlessly off the thioacetamide. In contrast, a water molecule that slams forcefully against the thioacetamide has more of a chance of driving away the sulfur atom, thereby producing acetamide and H2S. We might find that we could plot a curve of the probability of reaction as a function of the velocity of approach of the two molecules along a line connecting their centers.
Unfortunately, the kinds of observations we have been describing for a reaction are an unattainable dream. We must try to find out what is happening during a reaction in a more indirect way. Frequently the most that we can say about a proposed mechanism of reaction is that it is not incompatible with the data. There is always the lingering possibility that some other mechanism of reaction might explain the same data just as well. A classic example of this ambiguity is the reaction of H2 and I2. In 1893, Max Bodenstein, in Germany, studied the reaction:

This was the first comprehensive kinetic study of a reaction occurring in the gas phase. From that time until 1967, virtually every kinetics text and treatise used this reaction as the ideal example of a two-body collision mechanism. One gas molecule of H2 collides with a molecule of I2, they reshuffle atoms, and two molecules of HI are the result. But in 1967, J. H. Sullivan showed that this reaction does not take place by a two-body collision at all, but by a complicated chain reaction. We shall see later why the data measured before 1967 could be explained with equal ease with the two-body or a three-body model.
Not only are we unable to watch individual molecules, we cannot choose the orientation of the molecules upon collision. The best we can do is to estimate the probability of the molecules’ being suitably oriented and then modify our calculations of rates of reaction by a suitable factor. Such a correction sometimes is used and is called a steric factor.
Discovering the Process of Reactions and Crossed Molecular Beams
In a gas reaction or a reaction in solution, we cannot even choose the velocity of approach of the reacting molecules. The molecules in a sample of gas will have a distribution of velocities. We can shift the distribution of velocities by varying the temperature of the gas. As illustrated in Figure 3-11, in nitrogen gas the fraction of all the molecules having a velocity greater than some value such as 1000 m sec-1 increases as the temperature increases. At 273 K, only 0.44% of the N2 molecules have velocities of 1000 m sec-1 or greater; at 1273 K, 35% have this velocity or greater; at 2273 K, this fraction increases to 55%. However, nothing that we can do to the system will give us one specific velocity.
We can remove the velocity distribution for certain reactions by using the method of crossed molecular beams (Figure 22-2). Instead of reactions occurring between molecules dispersed in a solution or a gas, beams of molecules or ions are passed through one another in an evacuated chamber with negligible amounts of other molecules present. are used. A pair of wheels with spaced openings control the velocity of molecules. The beam sources typically are ovens that emit a stream of gas molecules, and electrostatic fields that accelerate ions]] The molecules in the crossed beams react with one another and are scattered from the beams. The products of the reaction, and the unreacted initial molecules, can be observed as a function of angle of scattering by using a movable detector mounted inside the chamber. This arrangement has the great advantage that the velocity selectors can limit the beam to molecules with velocities in a chosen small range. A knowledge of the products of the reaction as a function of angle of deflection or scatter provides much more information about the process of reaction. The orientation problem remains in a molecular-beam experiment, but one can imagine experiments in which this factor is controlled as well. Intense magnetic or electric fields placed just before the beams intersect might give the majority of the molecules in the beam one preferred orientation in space if the molecules had magnetic moments or dipole moments.
Some of the reactions that have been studied with crossed molecular beams are


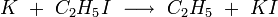
The reactants, beams of K atoms, HBr, CH3I, and C2H5I molecules, are emitted from heated ovens within the evacuated chamber. The detector is a heated wire filament called a surface ionization detector, which is sensitive to alkali metals or compounds of alkali metals.
Yuan T. Lee, a winner of the 1986 Nobel Prize in Chemistry, has extended molecular beam experiments to include larger and more complex molecules. He studied various reactions between organic molecules and fluorine or oxygen atoms. His most recent work has focused on basic reactions related to ozone depletion. Yuan Lee has utilized crossed-molecular beams to further explore the depletion of ozone by sending a beam of Helium seeded with ozone (O3) through a pulsed laser beam. The laser beam gives rise to high powered UV light and promotes the dissociation of O3 into a neutral O and O2. This research suggests that we could potentially control this sequence of reactions in order to create stratospheric O3 which would then be released into the environment, thereby repairing the Earth's ozone layer.[1]
Lee's group at the Lawrence Berkeley Laboratory is also using molecular beams to research photochemical processes. The researchers use a laser beam to excite molecules or atoms after they have been accelerated but before they collide, giving them more control over the type of chemical reaction that occurs. They are also studying the use of laser excitation during molecular beam experiments to promote the removal of one or more specific atoms from larger molecules as well as the combustion of hydrocarbons. [2]
Crossed-molecular beam research has facilitated a greater understanding of what intermediates (the short-lived molecules that form first in a molecular collision but soon decay into a more stable molecule) are formed in a reaction and how. By varying the velocities and angles of the incoming reactants, and by measuring and determining the distributions of the resulting products, researchers were able to infer the necessary and sufficient conditions under which the reaction would take place. In addition, the molecular cross beam has provided insight on how catalyses interact in chemical reactions. As more research is performed using crossed-molecular beam experiments, the ability to control the products of chemical reactions is becoming a reality. [3]
The disadvantage of molecular-beam experiments is that not all chemical reactions are suitable for study with molecular beams in evacuated chambers. Molecular-beam methods remain a special tool for making complete studies of certain special reactions. The majority of chemical reactions must be studied by bulk methods, in gas mixtures, solutions, and (less frequently) solids.
Measurement of Reaction Rates
The rate of reaction is usually followed in bulk methods by watching the disappearance of a reactant or the appearance of a product in a given time. If the chemical reaction is
- A + 2 B → 3 C
then the rate of appearance of product C in a time interval Δt is
![rate = \frac{\Delta [C]}{\Delta t}\,](https://upload.wikimedia.org/math/e/5/e/e5e6c0f71a1d8ce6a13b77278fbbe6c8.png) (22-1)
(22-1)
in which the concentration of C, [C], is usually expressed in moles liter-1. This is the average rate of appearance of C during the time interval Δt. The limit of this average rate as the time interval becomes smaller is called the rate of appearance of C at time t. It is the slope of the curve of [C] versus t at time t. This instantaneous slope or rate is written
Since one molecule of A disappears for every three of C that are produced, and two molecules of B disappear during the same process, the rates of disappearance and appearance of chemical species are related by the expression
The rate of a chemical reaction will depend on the concentrations of the reactants, although not always in the way that might be expected from the overall chemical equation. For the reaction of hydrogen gas with gaseous iodine to produce HI,
- H2 + I2 → 2 HI
the relationship between rates is
and as you might intuitively expect, the rate equation is
![\frac{d[HI]}{dt}\,=k[H_2][I_2]](https://upload.wikimedia.org/math/d/c/6/dc691b96d33e195cb3e0a65f37cc6627.png) (22-2)
(22-2)
The rate of reaction is proportional to the concentrations of H2 and I2, and dependent on the first power of each concentration. This does not mean that the reaction proceeds by a collision of one H2 molecule with one I2; since 1967 we have had evidence that it does not. We must distinguish clearly between the order of a reaction and the molecularity of the reaction. The constant of proportionality in equation 22-2 is called the rate constant.
The order of a reaction is the sum of all the exponents of the concentration terms in the rate equation. The HI reaction is first order in each of the reactant concentrations and is second order overall. Order is a purely experimental parameter and describes what is observed about the rate equation rather than implying anything about the mechanism of reaction.
The molecularity of a simple one-step reaction is the number of individual molecules that interact in the reaction. Molecularity requires a knowledge of the reaction mechanism. A reaction such as that of hydrogen and iodine actually may take place as a series of half a dozen individual reactions for which we could specify the molecularity of each. The concept of the molecularity of an overall reaction that occurs in a series of steps has no meaning. Most simple one-step reactions are unimolecular (spontaneous decay) or bimolecular (collision). True trimolecular reactions are rare, as three-body collisions are unlikely. Tetramolecular and higher reactions are virtually unheard of. Reactions that appear to be trimolecular or higher (from their stoichiometry) are, after careful study, usually seen to be the sum of a series of simple unimolecular and bimolecular steps. One of the challenges of chemical kinetics is to determine the true set of reactions in such a case.
The reaction of hydrogen gas with bromine is in complete contrast to that with iodine. The overall reaction is similar:
- H2 + Br2 → 2 HBr
but the experimental dependence of the rate of production of HBr upon concentrations of reactants and products is utterly different from equation 22-2:
![\frac{d[HBr]}{dt}\,=\frac{k[H_2][Br_2]^{1/2}}{1+k'([HBr]/[Br_2])}\,](https://upload.wikimedia.org/math/4/0/7/40755e38469dd2283afab410434c936f.png) (22-3)
(22-3)
This expression has two experimental rate constants, k and k’. We cannot talk about the molecularity of the reaction, because the overall process is the result of an elaborate chain of reactions that we shall come back to later. Even the order is a puzzle. At the start of a reaction of H2 with Br2, when little HBr is present, the second term in the denominator can be neglected. Then the reaction is effectively 11/2 order: first order in H2 and one-half order in Br2. As the product, HBr, accumulates, it slows down the rate of production of more HBr. Therefore, HBr is called an inhibitor of the reaction.
The formation of HCl is even more complicated. The production of HCl is accelerated by light of intensity I and is inhibited by the presence of oxygen gas, even at low oxygen concentrations. For years the difficulty of purifying the H2 and Cl2 gases and eliminating all traces of O2 led to erroneous conclusions about the kinetics of this reaction. The best experimental rate equation for the appearance of HCl is
![\frac{d[HCl]}{dt}\,=\frac{k_1I[H_2][Cl_2]}{k_2[Cl_2]+[O_2]([H_2]+k_3[Cl_2])}\,](https://upload.wikimedia.org/math/e/7/b/e7b026d78c8c9f65c8a4408e7d2f1ad1.png) (22-4)
(22-4)
Notice that, in the limit of the complete absence of oxygen gas, the rate is proportional to the concentration of H2 gas and not dependent on the concentration of Cl2 gas at all! (The second term in the denominator of equation 22-4 is zero, and the remaining Cl2 concentrations in the numerator and denominator cancel.) The reaction is complicated by side reactions that take place on the surfaces of the reacting vessels. The results obtained sometimes depend on the size and shape of the reaction container. All of this is a far cry from the simplicity of the HI system. There are side reactions in the HI system, too, but they are not important below 800 K.
Following the Course of a Reaction
How do we measure concentrations of reactants and products during a reaction to find rate equations such as the ones we have been examining? If the total number of moles of gas changes during a gas reaction, the course of the reaction can be measured in two ways: (1) the change in pressure at constant volume or (2) the change in volume at constant pressure. These are examples of physical measurements that can be performed on the system while it is reacting. They have the advantage of not disturbing the reacting system, and they are usually rapid. With automatic recording devices, we can monitor a physical quantity continuously during the reaction.
Other physical measurements often used in kinetic studies include optical methods such as the rotation of light by a solution (useful if reactants and products have different abilities to rotate polarized light), changes in refractive index of a solution, color, and absorption spectra. Common electrical methods include the electrical conductivity of a solution (especially useful when ions are being produced or consumed), electrical potential in a cell, and mass spectrometry. Thermal conductivity, viscosity of a polymerizing solution, heats of reaction, and freezing points also have been used. The disadvantage of all these methods is that they are indirect. The property observed must be calibrated in terms of concentrations of reactants and products. The calibration is subject to systematic errors, especially if there are side reactions occurring.
Several methods are used to prepare a system for observation in a kinetics experiment. One of today's common methods, called the 'temperature-jump' or 'T-jump' method, was developed in the 1950s by German physicist Manfred Eigen, who later shared the 1967 Nobel Prize in Chemistry for his work. In the T-jump method, a small sample of the system being studied is contained and allowed to react to equilibrium. Then the temperature of the system is raised several degrees in as little as 100 nanoseconds and the reaction is observed as it adjusts to its new equilibrium point. 100 nanoseconds turns out to be too slow for some reactions, but the T-jump method is adequate for a great number of chemical processes. The men with whom Eigen shared his prize, R.G.W. Norrish and George Porter, developed the flash-photolysis method, which was similar to the T-jump method in that it provided a way for making observations of high-speed reactions. Often scientists who excite reactions using the T-jump method observe using spectrometry; a 2009 study by Michael Frunzi, Hai Xu, R. James Cross, and Martin Saunders used NMR to determine the kinetics of the reaction of C60 with 9,10-dimethylantracene.
Chemical methods are more straightforward and yield concentrations directly. With such methods, a small sample is extracted from the reacting mixture, and the reaction is halted by dilution or cooling the mixture long enough to measure concentrations. A serious disadvantage is that we are removing a part of the reacting system and thereby gradually changing it. Moreover, if the reaction cannot be stopped in the sample that is removed for analysis, then the analysis is that much less accurate. In the gas-phase reactions between H2 and Cl2, Br2, or I2, there is no change in the number of moles of gas before and after reaction, so pressure- or volume-change methods cannot be used. To study these reactions, samples are taken, and the gas mixtures are analyzed chemically for their compositions.
A First-Order Rate Equation and the Decay of 14C
- Wikipedia article: Carbon-14
In a first-order process, the rate of disappearance of a reactant is proportional to the amount of the reactant present. Each reactant molecule has the same probability of breakdown in a given time interval; the total rate of breakdown simply depends on how many molecules are present. The expression is
with n being the total number of molecules present. This rate expression can be integrated to yield the concentration as a function of time:
This rate equation is used in the example of dating with 14C in section 23.5, where the expressions in terms of the concentration of 14C are
![\frac{d[^{14}C]}{dt}=-k[^{14}C]\,](https://upload.wikimedia.org/math/6/5/7/6574f2e924a5302490bb2aca8d003fa0.png) (22-5)
(22-5)
![[^{14}C]=[^{14}C]_0e^{-kt}\,](https://upload.wikimedia.org/math/f/4/b/f4b5fc7e6145d8b598de9c376de1feea.png) (22-6)
(22-6)
The integrated equation 22-6 is plotted in figure 23-3. Taking the logarithm of both sides of equation 22-6 yields
![ln[^{14}C]=[^{14}C]_0-kt\,](https://upload.wikimedia.org/math/e/3/b/e3b0f538820438fd14a6eeda746f3a40.png) (22-7)
(22-7)
This is the equation of figure 23.8. The plot is a straight line, with a negative slope equal to the rate constant, k. The slope of the plot of equation 22-6 at any time t, as shown in figure 23-3, is proportional to the concentration of 14C remaining at that time. This, in words, is what equation 22-5 means.
An alternative view While this is the traditional solution for the integration of the first order process, the use of Eulers constant in this situation introduces a systematic error in the reported kinetic constants.
As previously stated, in a first-order process, the rate of disappearance of a reactant is proportional to the amount of the reactant present.
Therefore the kinetic constant must represent the fraction of the population of reactant present that will breakdown in a given time period and the fraction must be less than one.
For rates that are very small in comparison to the total population the traditional equation works fairly well.
For example, for simplicity, if the initial population is assumed to be 1 (no=1) and we restrict the time period to the first interval (t=1), and we examine a rate of 5% conversion per time period we would assume a remainder of 95% of the original reactant after the first time period. However inserting these terms we get
To get the expected remainder of 95% this equation requires the kinetic constant to be increased to 5.129%.
The problem becomes more apparent the higher the rate observed, for example if the rate was 95%, then the remaining reactant after one time period would be expected to be 5% of the initial starting population however,
To produce the expected 5% remainder the rate constant must be increased to k=2.99573 a rate of approximately 300%, which may be difficult to observe.
The problems introduced by the use of this equation can be overcome by recognizing the artificial spliting of the constant as both eulers constant and the kinetic constant are constant and do not change, so a constant raised to a constant is a constant. This constant represents the fraction of the reactant population remaining (%RP) per time period so can also be rewritten to incorporate the rate of the fraction of the population that will breakdown (%BD) per time period as well.
Therefore the kinetic equation for first order kinetics can be rewritten as
Where the fraction of the population that will breakdown (%BD) can be expressed as the observed reaction rate (r) divided by the initial reactant concentration no.
This notation relates the kinetic constant directly to the observed rate and recognizes the kinetic constant can not exceed 1 as the rate can never be a value greater than the number in initial starting reactants.[4]
Decomposition of N2O5
 Table 22-1
Table 22-1In section 16.5, we encountered the decomposition of solid N2O5 as an example of a reaction that is spontaneous yet strongly endothermic. Now let's look at the decomposition of N2O5 dissolved in carbon tetrachloride as an example of a first-order chemical reaction. Solid N2O5 and one product, NO2, are soluble in CCl4; the other product, O2, is not. The reaction
- N2O5 → 2 NO2 + O2(g)
can be followed by measuring the total volume of oxygen gas that bubbles out of the solution.
The data for this reaction are given in Table 22-1, after O2 volume measurements have been converted to concentrations of N2O5 left in the solution. These data are plotted in Figure 22-3 as an example of the way in which concentration data are treated. The figure shows the concentration of N2O5 at any time, the rate of change in this concentration, and this rate of change divided by the concentration. This last quantity is equal to the rate constant. That the rate of change divided by the concentration is constant (within the limits of experimental error in the data in Table 22-1) demonstrates that the reaction is indeed first order.
Stoichiometry and Rate Expressions
 Figure 22-3
Figure 22-3The reaction
- 2 NO(g) + O2(g) → 2 NO2(g)
has an observed rate equation of the form
The reaction is second order in NO and first order in O2, and is third order overall. The rate equation happens to agree with the stoichiometry of the chemical reaction; this agreement suggests (but does not prove) that this may be a simple one-step reaction involving three molecules. In contrast, ethanol and decaborane react in solution according to the equation
- 30 C2H5OH + B10H14 → 10 B(OC2H5)3 + 22 H2
One might naively expect this to have a thirty-first order rate expression. In fact, the reaction is second order, first order in each of the two reactants. For the rate of disappearance of ethanol,
The Goals of Chemical Kinetics
Some chemical processes are simple one-step reactions involving one, two, or occasionally three molecules. Many more processes are the combination of several such simple reactions. One of the goals of chemical kinetics is to find out what the true molecular mechanism of a complex process is. Why do HI, HBr, and HCl have such different experimental rate equations for a reaction that looks superficially the same in all three cases? To a kineticist the question, “What is the mechanism of the reaction?” means this: “What is the sequence of simple reactions that produces the observed kinetics and stoichiometry of the overall reaction?” To this question organic and inorganic chemists have added, “What is the geometry of the reaction for each simple step in the overall process?” The goal of this inquiry is to predict why the simple reactions proceed as they do and to predict the rates at which they occur. The theories that have been developed to calculate the rate constants for simple unimolecular and bimolecular reactions are our next topic.
Calculating Rate Constants from Molecular Information
Let us assume a simple bimolecular reaction,
with a rate expression,
How far can we go in calculating k from the molecular properties of A, B,C, and D? One of the earliest observations was that k varies with temperature; the rate constant is larger, and the rate of reaction is faster, at higher temperatures.
Arrhenius’ Activation Energy
Arrhenius’ activation energy by definition is the energy that must be overcome in order for a chemical reaction to occur. The activation energy of a reaction is usually denoted in units of kilojoules per mole. The theory and history of Arrhenius activation energy will now be discussed. If we plot the logarithm of the rate constant against the reciprocal of temperature, we usually obtain a straight line. Although Arrhenius was not the first person to do this, he developed the idea and gave it an explanation. Therefore, such a plot is called an Arrhenius plot. What does it mean in terms of reaction mechanisms? Van’t Hoff and others had been working, in the late 1800s, on the variation of free energy change of reaction and of the equilibrium constant with temperature. They discovered that the equilibrium constant, Keq, varies with absolute temperature, T, and with the heat of reaction in the following way:
This expression can be derived from the Gibbs—Helmholtz equation and ultimately can be derived rigorously from thermodynamics. During the same period, G. M. Guldberg and P. Waage found that they could derive the equilibrium constant from kinetic arguments. If the forward reaction in the above general bimolecular reaction has the rate
and the reverse reaction has the rate
they assumed equilibrium was the state in which forward and reverse rates are equal, so no net change in the reacting system is occurring with time:
The equilibrium constant, in this argument, is the ratio of the rate constants for the forward and reverse reactions. This is an erroneous derivation. It is valid only when the reaction is a simple one-step process in which the stoichiometry of the reaction is reflected in the coefficients of the concentration terms in the rate equation. Nevertheless, it is valid for the kind of reactions we are considering here: simple bimolecular reactions. If the equilibrium constant is the ratio of forward and reverse rate constants, the above equation suggests that the enthalpy of reaction might be the difference between two energies, Eforward and Ereverse: G°=Eforward—Ereverse If the Arrhenius energy of activation is not a function of temperature, this equation predicts that a plot of In k against the reciprocal of the absolute temperature will generate a straight line. This is true for many reactions, but not all, and the activation energy is one of the standard experimental parameters by which a chemical reaction is described. A simple, but accurate, representation of the temperature dependence of the rate of a chemical reaction can be expressed in the following formula.
This expression can be rearranged to yield the more common Arrhenius equation.
A historically useful generalization supported by the Arrhenius equation is that, for many common chemical reactions at room temperature, the reaction rate doubles for every 10 degree Celsius increase in temperature. The activation energy is a barrier that the colliding molecules must surmount if they are to react rather than recoil from one another.
Using the reaction of water with thioacetamide we can postulate that if thioacetamide and water molecules do not collide head-on with sufficient energy the redistribution of bonds and subsequent reaction will never occur. Water will recoil from the thioacetamide molecule and no reaction will take place. This recoil is due to the fact that insufficient energy was provided to overcome the needed Arrhenius activation energy. With this established we have experimental evidence, in the form of the temperature dependence of k, that some such threshold energy is involved in chemical reactions.
Arrhenius’ explanation of activation energies assumes that every pair of molecules with energy less than Ea will not react, and every pair with energy greater than Ea will react. Nothing is changed in the thermodynamics of the overall reaction. The activation barriers to forward and reverse reactions, Eforward and Ereverse, are such that their difference, ΔH° = Eforward — Ereverse, is the thermodynamic heat of reaction. The higher the barrier, Eforward, the slower the forward reaction will be. However, since Ereverse must rise by the same amount as Eforward if their difference is fixed, the reverse reaction is slowed by the same amount.
The point of equilibrium is affected not by the individual numerical values of the activation energies for forward and reverse reactions, but only by the difference between them, which is ΔH°.
Collision Theory of Bimolecular Gas Reactions
The next logical step is to construct a collision theory for gas reactions. A reaction between two molecules occurs, in this theory, when the molecules collide with energy in excess of Ea. A theory could hardly be simpler. There are two questions to be answered before the rate constant can be calculated:
1. How often do two molecules collide per cubic centimeter of gas mixture?
2. In what fraction of these collisions does the combined energy of the two molecules exceed Ea?
The collision frequency can be calculated from the simple kinetic theory of gases with the methods that were introduced in Chapter 3. The frequency depends on the concentrations of the two reacting gases, and also on their molecular weights, the distance between the molecular centers on collision, and on the square root of temperature. Since the molecules move more rapidly at higher temperatures, they collide more often. The fraction of pairs of molecules having energy equal to or greater than Ea upon collision is
According to simple collision theory, the rate of reaction then is
The rate of a reaction is greater at higher temperatures because collisions are more frequent and because the probability that a colliding pair will have an energy greater than E is also higher. The constant, Z, can be calculated from the molecular weights and the diameters of the reacting molecules by approximating them with spheres. The bimolecular rate constant, k, then is
This theory is tested in the data in Table 22-2. The Arrhenius activation energy is tabulated for six bimolecular gas reactions, along with the observed pre-exponential factor, pz, and its theoretical value as calculated from the collision theory and the absolute rate theory that we will discuss in the next sections.
 Table 22-2
Table 22-2Keep in mind that these are the logarithms of Z that are tabulated, so a disagreement between theory and experiment of 1.0 means an error by a factor of 10 in the rate constant. The agreement is generally encouraging for so simple a theory that has no assumptions other than those of the kinetic theory of gases. There are discrepancies; for example, the ClO reaction rate is incorrect by a factor of 400. When discrepancies occur, the absolute rate theory usually does a better job of predicting Z than the collis ion theory does.
In the right column of Table 22-2 are the standard enthalpies or heats of reaction. The relative enthalpy of reactants and products, and the activ ation barrier between them, are plotted for these reactions in Figure 22-5. Some reactions, such as NO2 + CO, must surmount a considerable activat ion barrier. For other reactions, the barrier is nonexistent, as with 2Cl0. For others such as 2NO2, the barrier to reaction is only the heat of reaction itself, and the reverse reaction has a zero activation energy. The most general case is diagrammed at the bottom of Figure 22-5.
 Figure 22-5
Figure 22-5 Molecular Table
Molecular Table Figure 22-6
Figure 22-6Activated Complexes
Before we consider the absolute rate theory, we must look more closely at the state of the reactant molecules as they cross the activation barrier. In the reaction
the Cl and O atoms are bonded at the start, and the two ClO molecules are too far apart to exert any influence on one another. At the end of the reaction, the Cl atoms are 1.99 A apart in a Cl2 molecule, the O atoms are 1.21 A apart in an O2 molecule, and these two molecules are far apart. What is the intermediate, activated state?
The activated complex is diagrammed in Figure 22-6b. All four atoms are an unspecified distance apart, somewhat farther away than if they were bonding in the stable sense of the term. We can be sure that the activated complex is not one in which all four atoms are so far apart that they exert no influence on one another; some sort of a loose complex must exist. The basis for this assertion is a knowledge of the bond energies of the three molecules, and that the activation energy for the 2ClO reaction is zero. The bond energies, or the energies required to separate completely the atoms in a diatomic molecule, are shown in the molecular table.
If during the reaction the two ClO molecules were first pulled apart, and then the isolated atoms were combined into Cl2 and O2, the activation energy for this reaction would be twice the bond energy of ClO, or 540 kJ per 2 moles of ClO. Instead, the activation energy is zero. The activated complex must be a combination of the four atoms such that whatever instability created as Cl and O separate is immediately compensated by the stabilizing influence of associations between Cl and Cl and between O and O.
We can think of the activated complex as an unstable “molecule,” with many of the properties of a molecule, except that it decomposes spontaneously either to reactants or to products. The thioacetamide and water molecules in Figure 22-lb are in an activated complex, and the energy of this complex is greater than either that of thioacetamide and water or that of acetamide and hydrogen sulfide.
Potential Energy Surfaces
The 2ClO reaction suggests that in principle we should be able to calculate the total potential energy of a collection of atoms as a function of their positions in space. This calculation would produce a potential energy surface with hills and plateaus of high energy, and valleys of low energy. Any region of a minimum of potential energy in this plot will represent a stable molecule. Even with four atoms as in the 2Cl0 reaction we would need, unfortunately, six variables to describe the arrangement of atoms: the bond lengths from each atom to the other three, for example. Our potential energy plot would have to be in seven-dimensional space. This is difficult to visualize and impossible to construct. We need an example with only two variables so the map can be plotted in three-dimensional space. One of the first maps to be calculated, by Henry Eyring in 1935, is the potential energy surface for the reaction
in which all three atoms are constrained to lie on a straight line. The only variables are the distances from the central hydrogen atom to the other two, r1 and r2; r1 being the distance between the first two atoms, while r2 is the distance between the second and third atoms.
The potential energy of the three-atom system as a function of r1 and r2 is shown in Figure 22-7a.
 Caption 22-7
Caption 22-7 Figure 22-7
Figure 22-7 Figure 22-8
Figure 22-8The actual arrangements of the three atoms at the six numbered points marked in color are drawn in Figure 22-8. Sections through this potential energy surface at fixed values of r1 are shown in Figure 22-7b. If either r1 or r2 is large, the three hydrogen atoms exist as an H2 molecule and an iso1ated atom. The potential energy section at constant r1, for r1 greater than 3 A in Figure 22-7b, is the same as that for an isolated H2 molecule in Figure 12-2. As an atom approaches an H2 molecule from the right (points 1 and 2 of Figures 22-7 and 22-8), the first noticeable effect is an increase in the potential energy of the system of three atoms.
The incoming atom is repelled by the molecule, and a more stable situation results if the atom rebounds and moves away again. If the atom has enough kinetic energy to keep approaching the H2 molecule, it will begin to weaken the H—H bond in the H2 molecule.
At point 3, both outer atoms are slightly farther from the central one than a normal H — H bond length, but the potential energy of the system of atoms is 25 kJ /mole higher than that of isolated H2 and H. Point 3 is the activated complex for the reaction. The activated complex can decompose either to products or to reactants. There is no reason why the three atoms in the state of point 3 cannot return to point 1 as well as proceed to point 5. What is certain is that the activated complex is unstable and must decompose. Points 1 through 5 in Figure 22-7 are connected by a colored dashed line called a reaction pathway. If we plot potential energy along this pathway, an activation energy barrier curve, such as those in Figure 22-5, results. Notice that the reaction pathway at all times is a path along a valley between steep walls. It may take 25 kJ of energy to build the activated complex of point 3, but it takes over 400 kJ to separate the atoms as at point 6. Even with reactions of molecules so complicated that we cannot calculate or even plot their complete multidimensional potential energy surface such profiles are still useful.
Absolute Rate Theory
In the absolute rate theory, also known as transition-state theory, a reaction takes place when an activated complex breaks down into products. Therefore, the rate of reaction is the product of three factors:
1. The concentration of activated complexes per cubic centimeter
2. The rate of breakdown of individual complexes or their rate of passage over the activation energy barrier
3. The probability that a breakdown will form products and not reactants again
Since the activated complex represents an unstable state of transition between reactants and products, it is often called a transition state. The transition-state theory assumes an equilibrium between reactants and the activated complex, usually represented by a superscript double dagger:
Hence, the concentration of the activated complex is given by:
The rate of decomposition is more complicated to calculate, but it turns out to be a universal constant for all bimolecular reactions at a given temperature:
in which k = Boltzmann’s constant, and h = Planck’s constant. The probability that a breakdown will be to products and not to reactants is the transmission coefficient, m also often written as κ. It can be estimated only as having a value between 0.5 and 1.0 in most reactions. Therefore, the overall rate of reaction is
and the rate constant, k2, is
(The symbol k2 is used here instead of k to represent a bimolecular rate constant to avoid confusion with Boltzmann’s constant.) It is possible to calculate the equilibrium constant, K ‡, between reactants and the activated complex from molecular properties by looking at the thermodynamic interpretation of this rate-constant expression. The equilibrium constant is related to the standard free energy of formation of the activated complex from reactants, and this in turn is related to the standard enthalpy and entropy of the formation of the activated complex:
Thus, the bimolecular rate constant, k2, can be written
The enthalpy of activation, ∆Ho‡, is nearly the same quantity as the activation energy, Ea. The difference is irrelevant is this discussion. The equation above indicates that the rate of reaction is slower if the activation energy is large. This result was already obtained in the collision theory; if the activation energy is large, only a small fraction of the molecules will have enough energy to surmount the barrier and to react rather than rebound upon collision. The equation also suggests that the reaction rate is faster if the entropy of activation is large. If the activated complex is much more disordered than the reactants, the reaction is enhanced because the equilibrium constant for formation of the complex is large, and more of the complex is present. In contrast, if the reactants are severely constrained when they combine to make the activated complex, then the reaction is inhibited. We might guess that the entropy of activation in the thioacetamide-plus-water reaction is negative since the two molecules combine to form one unit in the complex. Both molecules are limited severely in their initial orientations if they are to build the activated complex successfully.
The entropy of activation in bimolecular reactions is almost always large and negative because the two reactants lose entropy when they combine in the complex. Often the most useful application of absolute rate theory is not to calculate the rate constant directly, but to use the observed rate constant and equation above to calculate the entropy of activation. The entropy of activation provides information about the structure of the activated complex. For example, if the calculated entropy of activation is positive, then any mechanism that leads through a tightly organized activated complex must be rejected. As an example, in the next section we will consider two reactions of the type:
which proceed by different mechanisms, depending on the nature of the R groups. In one mechanism, the Br- is driven away as OH- approaches, in the same fashion as the thioacetamide reaction. The activated complex is then a combination of R3C-Br and OH-. This is called an associative or SN2 reaction, meaning that it is a substitution of one group for another, that the groups are nucleophilic (donating electrons and attracting nuclei: Lewis bases, in fact), and that two molecules are involved in the reaction.
The other mechanism is for the R3C—Br molecule to dissociate spontaneously into Br and what is known as a carbonium ion, R3C, and for the OH- ions to react rapidly in a separate step with any free carbonium ions. The activated complex or transition state in this mechanism will be the reactant R3C—Br just before dissociation. This is called a dissociative or SN1 mechanism since it is a nucleophilic substitution in which the slowest step involves dissociation of a single molecule. One should be able to distinguish between these two mechanisms by their entropies of activation, which can be calculated from the equation above and the measured rate constants. The SN2 mechanism will have a large negative entropy of activation since the activated complex is formed by combining two molecules. In contrast, the SN1 mechanism will have virtually a zero entropy of activation because the activated complex differs only slightly from the reactant molecule.
Laser Control of Chemical Reactions
Given a set of molecules that can combine in two possible ways, scientists wonder how they can most effectively use a laser to prod the chemical reaction one way or the other. Because interference of molecular pathways is the key to governing reactions, any laser scenario that induces such interference may serve as a means of controlling reactions. Instead of shining two steady beams on a target, one might use ultrashort pulses of laser light. Modern lasers can generate bursts as short as 10-15 second. Unlike continuous-wave radiation, a light pulse is made up of a collection of distinct frequencies and, hence, of a collection of photons with different energies. Such light also has a perhaps counterintuitive property. The briefer the pulse, the broader the range of energies within it.
This property plays a major role in pulsed-laser methods for controlling the outcomes of chemical reactions. By delivering a range of energies, a pulse can induce motion (such as vibration or rotation) in a molecule, which in turn affects the way it interacts with other light pulses. Ordinarily a molecule exists at a specific energy value. A system at one of these fixed energies resides in a so-called stationary state and does not move over time. For a molecule to undergo the dynamics, it must live in several energy levels at once. Such an assemblage of energy levels is called a superposition state. The wave function describing the superposition state is the sum of wave functions representing stationary states of different energies. To construct it, researchers shine a pulse of coherent laser light on the molecule. The way the molecule then moves depends on the nature of the light pulse and its interaction with the molecule. Thus, we can affect dynamic changes in the molecule by shifting the relative contribution of the frequencies that compose the pulse--that is, by shaping the pulse.
Several researchers have developed these ideas. They include Stuart A. Rice of the University of Chicago, Robert J. Gordon of the University of Illinois at Chicago, Herschel Rabitz of Princeton University, Ronnie Kosloff of the Hebrew University of Jerusalem and Kent R. Wilson of the University of California at San Diego. Their results show that pulses built out of a complicated mixture of frequencies are required to control molecular dynamics optimally, but simple approximations often suffice to break apart molecules in a controlled way.
Complex Reactions
Most chemical reactions are not simple unimolecular or bimolecular reactions, but combinations of these. This is why such complicated rate equations as equations 22-3 or 22-4 arise. Even the hydrogen—iodine reaction, which has been used for over half a century as the classic example of a simple bimolecular reaction (equation 22-2), is complex.
The Hydrogen—Iodine Reaction
For the reaction
H2 + I2→ 2HI(22-19)the observed rate equation is
![-\frac{d[H_2]}{dt}\,=k[H_2][I_2]](https://upload.wikimedia.org/math/1/0/9/10936ca155d79223d8b8db7d0c7f1fb7.png) (22-20)
(22-20)However, both N. N. Semenov and Henry Eyring have suggested that the true mechanism might be not that of equation 22-19, but a two-step mechanism involving the reversible dissociationof I2 to 2I, followed by the trimolecular reaction of I and H2:
I2 ↔ 2I
H2 + 2I → 2HI(22-21)The rate expression for the reaction of one H2 molecule with two I atoms is
![\frac{d[H_2]}{dt}\,=k'[H_2][I]^{2}](https://upload.wikimedia.org/math/9/d/b/9dbb9ec0aec24e8144bae5d97d8b492d.png) (22-22)
(22-22)and if the dissociation of I2 is reversible and at equilibrium, we can write an equilibrium-constant expression:
![K = \frac{[I]^2}{[I_2]}\,](https://upload.wikimedia.org/math/a/0/6/a06b3c678b9e6e3f17681b2f70a3f801.png) [I]2=K[I2](22-23)
[I]2=K[I2](22-23)Then substituting the concentration of I atoms in equation 22-22 with the equilibrium equation 22-23 it produces
![-\frac{d[H_2]}{dt}\,=k'K[H_2][I_2]](https://upload.wikimedia.org/math/a/9/3/a9328cd04ee1380cde450d3c59900cf9.png) (22-24)
(22-24)which is the same rate expression as if the mechanism were really one of bimolecular collision. We thus have two different mechanisms with the same rate expression. How can we choose between them?.(Above 800 K, side reactions with different mechanisms occur, yet these reactions can be neglected at moderate temperatures.)
The two mechanisms have the same rate equation so long as the dissociation of I2 is at thermal equilibrium, and the amount of I atoms present is given by the thermal equilibrium constant of equation 22-23. At higher temperatures, more I2 dissociates, thereby producing the same effect that would have resulted from the greater bimolecular rate constant in the bimolecular mechanism. J. H. Sullivan decided to test the two mechanisms by making the concentration of iodine atoms different from what it is normally in the thermal dissociation of I2. He did this by dissociating I2 with 578-nm light from a mercury vapor lamp. This light should have had little effect if the reaction was bimolecular, aside from a slight decrease in the I2 concentration. Conversely, if the trimolecular reaction was correct, the rate of reaction should have increased with the intensity of irradiating light since more I atoms were being produced.
Sullivan calculated the concentration of I atoms present at several intensities of irradiating light and found that the rate of appearance of HI is proportional to the square of the I atom concentration. Therefore, the mechanism of equation 22-21 is the correct one. The classical H2 + I2 reaction is a trimolecular reaction imitating a simpler bimolecular reaction because of the thermal equilibrium that normally exists between I2 and 2I. (At least, until someone even more ingenious designs an experiment that proves that it is a more complicated reaction imitating a trimolecular reaction.) As Sullivan points out [j Chem. Phys. 46, 73 (1967)], the trimolecular reaction
H2+2I→2HIcan be replaced by two bimolecular steps:
H2+2I→H2I
H2I+I2↔HIIf the first of these is fast and reversible, so that reactants and products are in equilibrium, then the rate expression is the same as for the trimolecular process, and the two mechanisms cannot be distinguished by reaction rates. This example makes a point that must be kept in mind at all times - we can never prove that a proposed mechanism is right; we can only prove that it has not yet been shown to be wrong. There is always the chance that a more subtle experiment, such as Sullivan’s upsetting of thermal equilibrium with light, may uncover the weaknesses in an accepted mechanism. When two theories are presented on the same topic, the temptation (and usually the wiser choice) is to opt for the simpler one, until data compels you to do otherwise. But you should always be prepared to change your mind when new data demands it.
Rates and Mechanisms of Substitution Reactions
The reaction of tert-butyl bromide with OH—,
(CH3)3CBr + OH- ↔ (CH3)3COH + Br-(22-25)has the experimental rate expression
-![\frac{d[(CH_3)_3CBr]}{dt}\,=k[(CH_3)_3CBr]](https://upload.wikimedia.org/math/6/e/1/6e1522fef9751ac914ba5ebf40bab96a.png) (22-26)
(22-26)The rate does not appear to depend on the OH- concentration at all. In contrast, the similar reaction with a less highly substituted carbon atom in ethyl bromide,
CH3CH2Br + OH- ↔ CH3CH2OH + Br(22-27)has the rate expression that we might expect from the chemical equation:
![-\frac{d[CH_3CH_2Br]}{dt}\,=k[CH_3CH_2Br][OH^-]](https://upload.wikimedia.org/math/f/2/f/f2f830c15fe292e97817d76a37fd913f.png) (22-28)
(22-28)Why should these two similar reactions proceed by different mechanisms and have different rate equations? And how is it that the rate in equation 22-26 can be independent of concentration of one of the reactants?
Reaction 22-25 takes place by the SN1 mechanism the equation

The tert-butyl bromide first dissociates in a slow reaction, and than the carbonium ion that is formed reacts immediately with OH—. Whenever a process takes place by a series of rapid steps with one slower step, the overall rate of reaction will be controlled by the slow step. The rate in this SN1 reaction depends entirely on how fast the molecules of (CH3)3CBr decompose. The capacity of reacting carbonium ions with OH— far exceeds the amount of carbonium ions supplied by the dissociation of tert-butyl bromide. The total amount of OH- present is unimportant.
Why do reactions go with different mechanisms? TheSN2 scheme is possible for ethyl bromide because there is room for three substituents of the C atom (CH3 and two H), plus OH— and Br. Because of this the activated complex
-
is sterically possible.
In tert-butyl bromide,C4H9Br, on the other hand, the groups attached to the carbon atom (three CH3) are large enough that OH- and Br- cannot bind at the same time. The activated complex of the SN2 reaction is impossible, and no reaction can occur until a molecule of tert-butyl bromide spontaneously dissociates. The dissociated Carbonium Ion is then the ion to attack, either by Br- to form the reactants again or by OH— to form the product. If Br— is present only as a result of a previous reaction of tert-butyl bromide, its concentration is probably much smaller than that of OH—, and most of the carbonium ions will be converted to tert-butyl alcohol, (CH3)3COH.
In general, a rate expression that disagrees with the stoichiometry of the overall reaction is an indication that the reaction is proceeding by a series of steps. Then the problem is to find a set of steps including a slow step that accounts for the observed rate law.
In the reaction mechanisms of tert-butyl bromide and ethyl bromide the rate difference is also encountered in the octahedral and square planar
complexes of the transition metals. The square planarcomplexes of Pt(II) and other metals can react with new ligands by associative (SN2) mechanisms because the metal atom is accessible from either side of the plane. The SN2 mechanism of the reaction
Pt(NH3)3Cl+ + Br- → Pt(NH3)3Br+ + Clcan be written
-
The activated complex is a five-coordinated platinum, which breaks down rapidly to products. The rate of the overall reaction depends on the rate of formation of the activated complex. This rate is influenced strongly by the nature of the entering group (Br- in this example). Ligands capable of forming strong bonds with the central atom are the best entering groups because they displace the leaving group (C1 in this example) rapidly. The ions CN— and I- are good entering groups for Pt(II) complexes, whereas NH3 and H20 are relatively poor.
It is much more difficult for the six-coordinated octahedral complexes to react by an SN2 mechanism because six ligands around a central metal, such as Co(III), leave little or no room for the attachment of an entering group in a transition state. Studies of substitution reactions of octahedral Co(III) complexes have established that the rate-determining step involves the dissociation of the bond between the Co(III) and the leaving group (the entering group is not involved in this dissociation step.) In aqueous solution, for example, H20 displaces Cl -in the complex Co(NH3)5Cl2+, thereby producing Co(NH3)5H203+. The mechanism that is consistent with the experimental rate of this and similar reactions is the dissociative or SN1 mechanism, which can be written.
-[[
For such mechanisms, the entering group plays no significant role in the creation of the Transition state and the rate of the overall reaction. A characteristic of most octahedralsubstitution reactions is the lack of influence of entering groups on the rate of reaction.
Chain Reactions
The reaction H2 + Br2 → 2HBr has the strange rate equation that we have already seen,
(22-3)For 13 years after this rate law was discovered, no one could account for it. Then, three groups scientist those groups of Henry Eyring, K. F. Herzfeld,and Michael Polanyidid so simultaneously. They proposed that the reaction proceeds by a chain mechanism involving two chain-propagating steps:
(1) H2+Br→ H+HBr (k1)
(2) H+ Br2 → HBr+Br (k2)When a molecule breaks apart into uncharged fragments having unpaired electrons, the unpaired electrons (e.g., in H and Br) make the fragments called radicals,chemically reactive. The atomic product of each of these steps is a reactant for the other step, and they both produce HBr. Thus, HBr results, not from a bimolecular collision, but from an endless chain of reactions 1 and 2. The first of these two steps is the reaction of Figure 22-9. But where do these atoms of Br and H come from? The Br atoms are postulated to come initially from a chain-initiating step:
(3) Br2 → Br+ Br (k3)Why is the dissociation of H2 not included also? The real reason is that the explanation of the reaction rate (equation 22-3) does not require it, and if we add it, we obtain the wrong rate expression. We can justify this omission in another way: The dissociation energy of H2 is 432 kJ mole-1, whereas that of Br2 is only 190 kJ mole-1. A large concentration of HBr inhibits the reaction ,as we can see from the HBr term in the denominator of equation 22-3, and a large concentration of Br2 counteracts this inhibition. From this it is evident that HBr and Br2 are competing for the same chemical substance. What might that substance be? The most likely candidate is hydrogen atoms and the inhibiting reaction would be
(4) H+HBr→H2+Br (k4)This is a chain-inhibiting reaction and is counteracted if an excess of Br2 makes reaction 2 go rapidly, as the rate equation 22-3 predicts. Finally, the chain is terminated by the recombination of Br:
(5) Br+Br → Br2(k5)If we can obtain equation 22-3 from these five reactions this will be a strong argument for the correctness of this chain mechanism (although not an absolute proof, as we have seen with HI.) The rate of appearance of HBr is given by
![\frac{d[HBr]}{dt}\,=+k_1[H_2][Br]+k_2[H][Br_2]-k_4[H][HBr]](https://upload.wikimedia.org/math/9/d/c/9dc0f8d966369a973e1bf809c64b8b96.png) (22-29)
(22-29)since HBr appears as a result of reactions 1 and 2, and disappears in reaction 4. The rates of production of H and Br atoms are given by
![\frac{d[H]}{dt}\,=k_1[H_2][Br]+k_2[H][Br_2]-k_4[H][HBr]](https://upload.wikimedia.org/math/1/c/4/1c42410e7198aadfe94d95fc9dcdb7ab.png) (22-30)
(22-30)![\frac{d[Br]}{dt}\,=-k_1[H_2][Br]+k_2[H][Br_2]+k_4[H][HBr]+2k_3[Br_2]-2k_5[Br]^{2}](https://upload.wikimedia.org/math/9/1/0/910864e2d58637589bc8d73ddf95bab8.png) (22-31)
(22-31)The coefficients of 2 in front of k3 and k5 arise because each unit of reaction 3 produces two Br atoms, and each unit of reaction 5 removes two Br atoms.
At this point an essential simplification must be made. It is assumed that the actual amount of H and Br atoms present at any time must be small because they are consumed at almost the same rate that they are produced. Soon after the reaction begins, the concentrations of H and Br will reach a steady state and will remain constant so long as the reaction continues with a plentiful supply of reactants. Because of this each of the rate equations in 22-28 and 22-29 can be set equal to zero:
![0=k_1[H_2][Br]-k_2[H][Br_2]-k_4[H][HBr]](https://upload.wikimedia.org/math/8/0/4/8047f6bdb56ee9e0dbb11402d7a2f484.png) (22-32)
(22-32)![0=-k_1[H_2][Br]+k_2[H][Br_2]+k_4[H][HBr]+2k_3[Br_2]-2k_5[Br]^{2}](https://upload.wikimedia.org/math/8/8/1/881083fe43ef8d65d7531828c6092a43.png) (22-33)
(22-33)Adding these two equations yields
![2k_5[Br]^{2}=2k_3[Br_2]](https://upload.wikimedia.org/math/3/6/3/36316b3c23c305b6c6fee65a973a7419.png)
with the rate equation
![[Br]=(\frac{k_3}{k_5}\,)^{1/2}[Br_2]^{2}](https://upload.wikimedia.org/math/c/1/3/c13696968c26fb4486b9b14c84ac12e3.png) (22-34)
(22-34)This calculation gives us a steady-state concentration for Br atoms in terms of the concentration of Br2 molecules. The HBr rate equation can be rewritten as
![\frac{d[HBr]}{dt}\,=k_1[H_2][Br]+(k_2[Br_2]-k_4[HBr])[H]](https://upload.wikimedia.org/math/a/8/b/a8b1c459f5b015b9e5c59b15ed80f2e4.png) (22-35)
(22-35)We can eliminate the H concentration by expressing it in terms of Br concentration from equation 22-32:
![[H]=\frac{k_1[H2]}{k_2[Br_2]+k_4[HBr]}\,[Br]](https://upload.wikimedia.org/math/2/b/6/2b6ce90e110aaf544013d9532ea0f996.png) (22-36)
(22-36)Substituting equation 22-36 into 22-35, placing everything over a common denominator, and canceling terms yields
![\frac{d[HBr]}{dt}\,=\frac{2k_1k_2[H_2][Br_2][Br]}{k_2[Br_2]+k_4[HBr]}\,](https://upload.wikimedia.org/math/7/a/c/7acdf6a1c9b09c9a04e016d35aa0fb73.png) (22-37)
(22-37)Dividing top and bottom by [Br2] and then eliminating [Br] with equation 22-34 yields
![\frac{d[HBr]}{dt}\,=\frac{1}{1+(k_4/k_2)([HBr]/[Br_2])}\,](https://upload.wikimedia.org/math/0/8/2/08206436ed10964e8f26e341d763895f.png) * (2k1(k3/k5)1/2[H2][Br2]1/2)(22-38)
* (2k1(k3/k5)1/2[H2][Br2]1/2)(22-38)This is exactly the experimental rate law, in which the experimental rate constants are related to those for the individual reactions in the chain by
k = 2k1 (k3/k5)1/2
k'= k4/k2Now that we know what these two experimental constants mean in terms of the individual reactions, we can give a much fuller interpretation to the rate law, equation 22-38. Suppose that we could vary the individual rate constants, k1 to k3, at will. What effects would these changes have on the overall rate? The overall rate of production of HBr is accelerated if rate constants k1, k2, and k3 are large, or if reactions 1, 2, and 3 are fast. The first two of these reactions produce HBr; the third prepares the way by making more Br atoms. The production of HBr is slowed if k4 and k5 are large, or if the chain-inhibiting and chain-terminating reactions are fast.
So long as k3 and k5 change together, there is no change in the overall rate of reaction. Reactions 3 and 5 are the opposing initiating and terminating steps. Similarly, so long as k2 and k4 change together, the rate is unaffected. This, too, is sensible; for reactions 2 and 4 are similar in that they both consume an H and produce a Br, but differ in producing HBr in reaction 2 and removing it in reaction 4. Inhibition by HBr occurs because reaction 4 is enhanced, and inhibition is lessened by Br2 because reaction 2 is enhanced.Practical uses of Complex Reactions
Although the chemistry behind complex reaction is not always understood, or even thought about, these reactions are used very often in the world around us.
A prime example of this is Hydrogen-Iodide which is used as a reactant in the complex reaction resulting in the production of methamphetamine. The production of methamphetamine utilizes the reduction of Ephedrine with Red Phosphorous, and Hydrogen-Iodide (aka Hydriodic Acid) [5]. The reaction mechanism for the reduction of Ephedrine with Red Phosphorous, and Hydrogen-Iodide is summarized as follows: Ephedrine reacts with Hydrogen-Iodide to form iodoephedrine (iodomethamphetamine) which is then reduced to methamphetamine [6]. The reduction of Ephedrine with Red Phosphorous, and Hydrogen-Iodide to create methamphetamine involves a cyclic oxidation of the iodide anion to iodine and reduction of iodine back to the anion by the red phosphorus [7]. This multi-step process is a classic complex reaction in which two reactants undergo an oxidation reaction to from an intermediate product. This intermediate chemical undergoes a reduction reaction with a third chemical, and transforms one of the original reactants back to its original state as well as changing the intermediate into the final product.
Classically, methamphetamine has been thought of as an illicit substance that used harsh chemicals to release a cascade of dopamine from the brain [8]. Although it is true that methamphetamine can be abused in this way, methamphetamine is also commonly used in medicine as well. In controlled dosages, and in specially designed time-release pills methamphetamine and Amphetamine are used to treat conditions such as Attention Deficit Hyperactivity Disorder, Traumatic Brain Injury, and the daytime drowsiness symptoms of Narcolepsy and Chronic Fatigue Syndrome. [9]. A typical daily dose of oral methamphetamine for the treatment of attention-deficit hyperactivity disorder in children is 20–25 mg. In the brain methamphetamine elevates the levels of extracellular monoamine neurotransmitters (dopamine, serotonin, norepinephrine) by promoting their release from the nerve endings. It is not completely understood how methamphetamine causes neurotransmitter release, but it appears to involve changing the distribution of the monoamine neurotransmitters from synaptic vesicles to the neuronal cytoplasm. It also reverses the transport of neurotransmitters from within the cells plasma membrane into the extracellular space [10]. This change in body chemistry results in the following symptoms and effects: alertness, wakefulness, energy, well-being, euphoria (at high doses) and suppression of appetite. Methamphetamine also activates the cardiovascular system (increased heart rate and blood pressure) and, for this reason, can cause death at high doses [11].
Another way in which complex reactions, and more specifically chain reactions, are used is in Nuclear Fission reactions. In these reactions an atom is bombarded by a neutron which splits the atom into two atoms which have a combined weight less than that of the original atom.[12] This reaction releases several products: 1) two smaller atoms; 2) heat; and 3) more neutrons. This allows the reaction to not only continue, but to grow in size on an exponential scale, thus becoming a chain reaction. This is the concept behind nuclear reactors and nuclear explosions; a sustained nuclear chain reaction of molecules that produces energy. For this reaction to occur and be efficient, atoms with high molecular weight, such as Plutonium and Uranium, must be used because their Binding Energy is low. As a result, when the atom is split, the amount of energy released is large. Also there is a specific mass, known as the critical mass, which is the mass the reactants must cumulatively be for the reaction to remain sustainable. If all of these criteria are met then a fission reaction will occur and it will result in a release of energy. The difference between a fission reactor and a fission bomb is the rate at which the fission reaction takes place.[13] In a nuclear reactor the metal is in a moderator, which slows the speed of the reaction, and the reaction is dispersed by control rods which absorb a portion of the free neutrons and slow the reaction further.[14] In these fission reactions the moderator, usually water, is used to make steam which powers a turbine and produces electrical power.[15]
References
- ALSNews,Ozone Photodissociation Probed Using Undulator Light,http://www.als.lbl.gov/als/als_news/...35_090595.html
- Gale Group Chemistry:probing reaction dynamics,en.wikibooks.org/w/index.php?...edit§ion=5
- Yuan T. Lee's Crossed Molecular Beam Experiment , http://www.osti.gov/accomplishments/YuanLee_Exp.pdf
- Walsh R, Martin E, Darvesh S. A method to describe enzyme-catalyzed reactions by combining steady state and time course enzyme kinetic parameters. Biochim Biophys Acta. 2010, 1800 (1), pp. 1-5
- Skinner, Henry F., "Methamphetamine Synthesis Via HI/Red Phosphorous Reduction of Ephedrine," Forensic Science International, 48 128-134 (1990)
- Skinner, Henry F., "Methamphetamine Synthesis Via HI/Red Phosphorous Reduction of Ephedrine," Forensic Science International, 48 128-134 (1990)
- Skinner, Henry F., "Methamphetamine Synthesis Via HI/Red Phosphorous Reduction of Ephedrine," Forensic Science International, 48: 128-134 (1990)
- Kish, Stephen J., "Pharmacologic mechanisms of crystal meth," CMAJ 13: 178 (June 17, 2008)
- Kish, Stephen J., "Pharmacologic mechanisms of crystal meth," CMAJ 13: 178 (June 17, 2008)
- Kish, Stephen J., "Pharmacologic mechanisms of crystal meth," CMAJ 13: 178 (June 17, 2008)
- Kish, Stephen J., "Pharmacologic mechanisms of crystal meth," CMAJ 13: 178 (June 17, 2008)
- Hill, John W., and Ralph H. Petrucci. General chemistry an integrated approach. Ed. Paul F. Corey. Upper Saddle River, N.J: Prentice Hall, 1999.
- Hill, John W., and Ralph H. Petrucci. General chemistry an integrated approach. Ed. Paul F. Corey. Upper Saddle River, N.J: Prentice Hall, 1999.
- Hill, John W., and Ralph H. Petrucci. General chemistry an integrated approach. Ed. Paul F. Corey. Upper Saddle River, N.J: Prentice Hall, 1999.
-
- Hydroformylation is the process of producing aldehydes from alkenes by adding a formyl group (CHO) and a hydrogen atom to a carbon-carbon double bond using homogeneous catalysis. It is particularly useful because the aldehydes that are formed can be converted into useful products such as detergents and specialty chemicals.[3]
- Ziegler-Natta catalysts are catalysts often based on titanium compounds and organometallic aluminium compounds. They are mostly used to polymerize terminal 1-alkenes.[4]
- The majority of today's knowledge about homogeneous catalysis comes from earlier studies of hydrogenation. Hydrogenation (as shown in the figure below) is a chemical reaction that results from bonding hydrogen to organic compounds through the use of catalysts.

-
The homogeneous catalysts involved in this reaction include Wilkinson’s catalyst, a rhodium-based compound, and Crabtree’s catalyst, an iridium-based compound. Moreover, many modern applications of hydrogenation can be found in petrochemical, pharmaceutical and food industries. [5]Carvone hydrogenation
- Carbon-hydrogen bond activation or CH activation may be defined as a reaction that forms a carbon-hydrogen bond. Although they are traditionally unreactive, CH bonds can form by coordination using a catalyst. A significant role of CH activation is the ability to convert inexpensive and abundant alkanes into valuable functionalized organic compounds. [6]
- In competitive reversible inhibition the substrate and the inhibitor compete for the enzyme's active site since they cannot bond to it at the same time. This type of inhibition can be overcome by high concentrations of substrate.[14] Competitive reversible inhibition can also occur when the competition is for the allosteric site instead of the active site.[15]
- In uncompetitive reversible inhibition the inhibitor binds to the complex created by the substrate binding to the enzyme's active site. Because the active site is no longer available, the binding efficiency decreases as does the maximum velocity of the enzyme.[16]
- In mixed inhibition the inhibitor can bind to the enzyme and its substrate simultaneously. While this effect can be reduced by a high concentration of substrate is cannot be completely overcome. This type of simultaneous binding can occur at the enzyme's active site, but generally occurs via the allosteric effect (The inhibitor binds to a site other than the active site.) which changes the shape of the enzyme and reduces the affinity of the substrate for the active site.[17]
- Non-competitive inhibition, a type of mixed reversible inhibition, reduces the activity of the enzyme without affecting the binding capabilities of the substrate. The extent to which inhibition occurs is directly dependent on the concentration of the inhibitor.[18] The inhibitor always binds to an allosteric site.[19]
- Bryant, Charles W. and Karim Nice. "How Catalytic Converters Work". http://auto.howstuffworks.com/catalytic-converter2.htm.
- Brittanica Online Encyclopedia, "Homogeneous Catalysis", http://www.britannica.com/EBchecked/topic/270491/homogeneous-catalysis
- Wikipedia, "Hydroformylation", en.Wikipedia.org/wiki/Hydroformylation
- Wikipedia, "Ziegler-Natta Catalysts",en.Wikipedia.org/wiki/Ziegler-Natta
- Wikipeida, "Hydrogenation", en.Wikipedia.org/wiki/Hydrogenation
- Wikipedia, "C-H Activation", en.Wikipedia.org/wiki/C-H_activation
- R. E. Dickerson, H. B. Gray, and G. P. Haight, Jr., Chemical Principles, 3rd edition, The Benjamin/Cummings Publishing Company, Menlo Park, CA, 1979, http://resolver.caltech.edu/CaltechBOOK:1979.001
- R. E. Dickerson, H. B. Gray, and G. P. Haight, Jr., Chemical Principles, 3rd edition, The Benjamin/Cummings Publishing Company, Menlo Park, CA, 1979, http://resolver.caltech.edu/CaltechBOOK:1979.001
- Accepta: Advanced Environmental Technologies, "Cooling Tower Scale & Corrosion Inhibitors" http://www.accepta.com/cooling-towers-water-treatment/cooling-tower-scale-corrosion-inhibitors.asp
- NCI Cancer Bulletin, "Aromatase Inhibitors Come of Age," vol. 4/no. 10, 6 March 2007 www.cancer.gov/cancertopics/t...inhibitors0307
- Protease Inhibitors: A Simple FactSheet from the AIDS Treatment Data Network, 15 Aug. 2006 http://atdn.org/simple/protease.html
- Consumer Reports: Best Buy Drugs www.consumerreports.org/healt...ager_ACEIs.pdf
- Wikipedia, Enzyme inhibitor, en.Wikipedia.org/wiki/Enzyme_inhibitor
- Wikipedia, Enzyme inhibitor, en.Wikipedia.org/wiki/Enzyme_inhibitor
- Wikipedia, Non-competitive inhibition, en.Wikipedia.org/wiki/Non-competitive_inhibition
- Wikipedia, Enzyme inhibitor, en.Wikipedia.org/wiki/Enzyme_inhibitor
- Wikipedia, Enzyme inhibitor, http://en.Wikipedia.org/wiki/Enzyme_inhibitor
- Wikipedia, Enzyme inhibitor, http://en.Wikipedia.org/wiki/Enzyme_inhibitor
- Wikipedia, Non-competitive inhibition, http://en.Wikipedia.org/wiki/Non-competitive_inhibition
![\frac{d[C]}{dt}.](https://upload.wikimedia.org/math/c/d/6/cd677dc435be9edaa104d012bee0b8d5.png)
![-\frac{d[A]}{dt}\,=-\frac{1}{2}\,\frac{d[B]}{dt}\,=+\frac{1}{3}\,\frac{d[C]}{dt}\,](https://upload.wikimedia.org/math/2/5/b/25bff69019e21da74191a0f29153d384.png)
![\frac{d[HI]}{dt}\,=-2\frac{d[H_2]}{dt}\,=-2\frac{d[I_2]}{dt}\,](https://upload.wikimedia.org/math/9/c/b/9cbda66348aed6ff8755b6963af301ea.png)



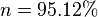




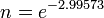




![\frac{d[NO_2]}{dt}=k[NO]^2[O_2]\,](https://upload.wikimedia.org/math/e/1/f/e1f968560ab4a54cf4b8f888ccfd6db0.png)
![-\frac{d[C_2H_5OH]}{dt}=k[C_2H_5OH][B_{10}H_{14}]\,](https://upload.wikimedia.org/math/f/2/f/f2f01d82787bea7781134ba0efb67082.png)

![\frac{-d[A]}{dt} = k[A][B]](https://upload.wikimedia.org/math/4/2/9/4297f466ae4a8089cdf01dc971f61085.png)

![Rate1\ = k_1[A][B]](https://upload.wikimedia.org/math/b/8/0/b808365b50211c183d9cd3061d2e263e.png)
![Rate2\ = k_2[C][D]](https://upload.wikimedia.org/math/5/5/3/553bc1907e46b190cb688bb4654fb22e.png)
![[A][B]k_1\ = k_2[C][D]](https://upload.wikimedia.org/math/e/a/4/ea4d3c9df7377662b36afbcaba8ccf9f.png)
![K_{eq}\ = \frac{k_1}{k_2}\ = \frac{[C][D]}{[A][B]}](https://upload.wikimedia.org/math/2/9/6/296d5020f7d7c031d59ab3224ba29dff.png)




![-\frac{d[A]}{dt}=(Z[A][B])(e^{\frac{-E_a}{RT}})](https://upload.wikimedia.org/math/3/4/1/3414b62d6f0478486da43c9a8cbb88d2.png)
![-\frac{d[A]}{dt}=Ze^{\frac{-E_a}{RT}}[A][B]](https://upload.wikimedia.org/math/a/0/a/a0afa047e19560fdb2d3ea4a21b94d31.png)





![K^{\Dagger} =\frac{[AB^{\Dagger]}}{[A][B]}](https://upload.wikimedia.org/math/5/e/6/5e63b7fe7e5f50207ea4d2445b923a35.png)
![[AB^{\Dagger}]=K^{\Dagger} [A][B]](https://upload.wikimedia.org/math/5/c/d/5cd502fb7d8ecb9e3fd5bcdb4e86daf9.png)

![-\frac{d[A]}{dt}=\kappa\frac{kT}{h}K^{\Dagger}[A][B]](https://upload.wikimedia.org/math/7/f/e/7fe5afa5f892270bd6c3d12fcf33b3b4.png)