
8.3: Fluorescence and Phosphorescence
- Page ID
- 190054

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)Sometimes, when an excited state species relaxes, giving off a photon, the wavelength of the photon is different from the one that initially led to excitation. When this happens, the photon is invariably red-shifted; its wavelength is longer than the initial one. This situation is called "fluorescence".
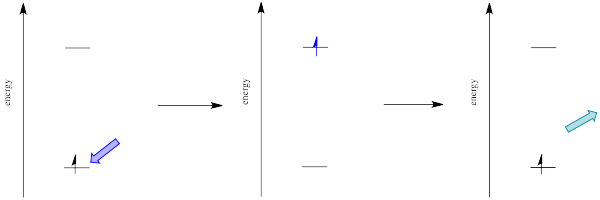
How can that be? Isn't energy quantized? How is the molecule suddenly taking a commission out of the energy the original photon brought with it?
Relaxation and Fluorescence
This discrepancy is related to the Franck-Condon principle from the previous page. When an electron is promoted to an electronic excited state, it often ends up in an excited vibrational state as well. Thus, some of the energy put into electronic excitation is immediately passed into vibrational energy. Vibrational energy, however, doesn't just travel in photons. It can be gained or lost through molecular collisions and heat transfer.
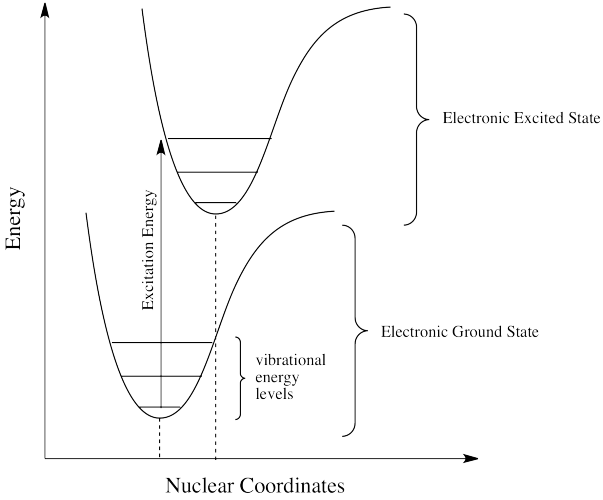
The electron might simply drop down again immediately; a photon would be emitted of exactly the same wavelength as the one that was previously absorbed. On the other hand, if the molecule relaxes into a lower vibrational state, some of that initial energy will have been lost as heat. When the electron relaxes, the distance back to the ground state is a little shorter. The photon that is emitted will have lower energy and longer wavelength than the initial one.
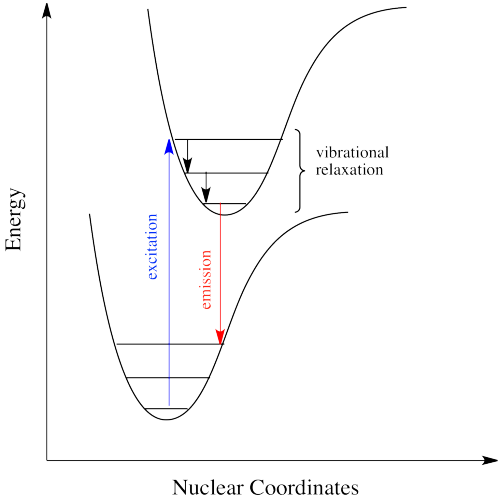
Just how does a molecule undergo vibrational relaxation? Vibrational energy is the energy used to lengthen or shorten bonds, or to widen or squeeze bond angles. Given a big enough molecule, some of this vibrational energy could be transferred into bond lengths and angles further away from the electronic transition. Otherwise, if the molecule is small, it may transfer some of its energy in collisions with other molecules.
There are lots of examples of energy being transferred this way in everyday life. In a game of pool, one billiard ball can transfer its energy to another, sending it toward the pocket. Barry Bonds can transfer a considerable amount of energy through his bat into a baseball, sending it out of the park, just as Serena Williams can send a whole lot of energy whizzing back at her sister. In curling, one stone can transfer its energy to another, sending it out of the house and giving Canada the gold over Sweden.
Exercise \(\PageIndex{1}\)
How does the energy of an electronic absorption compare to other processes? To find out, you might consider the excitation of an entire mole of molecules, rather than a sinle molecule absorbing a single photon. Calculate the energy in kJ/mol for the following transitions.
- absorbance at 180 nm (ultraviolet)
- absorbance at 476 nm (blue)
- absorbance at 645 nm (red)
- Answer a
-
a) E = h c / λ
E = (6.625 x 10-34 J s mol-1 x 3.0 x 108 m s-1 )/ (180 nm x 10-9 m nm-1) = 1.10 x 10-18 J
That's for one molecule. On a per mole basis, E = 1.10 x 10-18 J x 6.02 x 1023 mol-1 = 665,000 J mol-1 = 665 kJ mol-1.
For comparison, the relatively strong and unreactive C-H bond in methane has a bond dissociation energy of only 440 kJ mol-1. (That's a thermodynamic value; to actually break the bond would cost more input of energy, to get over the kinetic energy barrier for bond cleavage.)
- Answer b
-
b) E = h c / λ
E = (6.625 x 10-34 J s mol-1 x 3.0 x 108 m s-1 )/ (476 nm x 10-9 m nm-1) = 4.17 x 10-19 J
That's for one molecule. On a per mole basis, E = 4.42 x 10-19 J x 6.02 x 1023 mol-1 = 251,000 J mol-1 = 251 kJ mol-1.
- Answer c
-
c) E = h c / λ
E = (6.625 x 10-34 J s mol-1 x 3.0 x 108 m s-1 )/ (645 nm x 10-9 m nm-1) = 3.08 x 10-19 J
That's for one molecule. On a per mole basis, E = 3.08 x 10-19 J x 6.02 x 1023 mol-1 = 138,000 J mol-1 = 138 kJ mol-1.
Exercise \(\PageIndex{2}\)
How does the energy of an excitation between vibrational states compare to that of an electronic excitation? Typically, infrared absorptions are reported in cm-1, which is simply what it looks like: the reciprocal of the wavelength in cm. Because wavelength and frequency are inversely related, wavenumbers are considered a frequency unit. Calculate the energy in kJ/mol for the following transitions.
- absorbance at 3105 cm-1
- absorbance at 1695 cm-1
- absorbance at 963 cm-1
- Answer a
-
a) λ = 1 / wavenumber = 1 / 3105 cm-1 = 3.22 x 10-4 cm x 0.01 m cm-1 = 3.22 x 10-6 m
E = h c / λ
E = (6.625 x 10-34 J s mol-1 x 3.0 x 108 m s-1 )/ (3.22 x 10-6 m) = 6.17 x 10-20 J
That's for one molecule. On a per mole basis, E = 6.17 x 10-20 J x 6.02 x 1023 mol-1 = 37,000 J mol-1 = 37 kJ mol-1.
- Answer b
-
b) λ = 1 / wavenumber = 1 / 1695 cm-1 = 5.90 x 10-4 cm x 0.01 m cm-1 = 5.90 x 10-6 m
E = h c / λ
E = (6.625 x 10-34 J s mol-1 x 3.0 x 108 m s-1 )/ (3.22 x 10-6 m) = 3.37 x 10-20 J
That's for one molecule. On a per mole basis, E = 3.37 x 10-20 J x 6.02 x 1023 mol-1 = 20,000 J mol-1 = 20 kJ mol-1.
- Answer c
-
c) λ = 1 / wavenumber = 1 / 963 cm-1 = 1.04 x 10-3 cm x 0.01 m cm-1 = 1.04 x 10-5 m
E = h c / λ
E = (6.625 x 10-34 J s mol-1 x 3.0 x 108 m s-1 )/ (1.04 x 10-5 m) = 1.91 x 10-20 J
That's for one molecule. On a per mole basis, E = 1.91 x 10-20 J x 6.02 x 1023 mol-1 = 11,500 J mol-1 = 11.5 kJ mol-1.
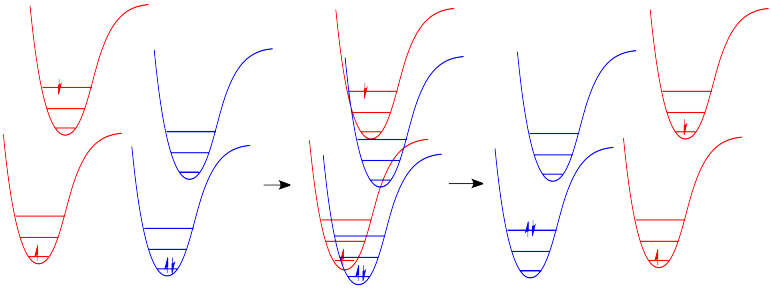
In molecules, as one molecule drops to a lower vibrational state, the other will hop up to a higher vibrational state with the energy it gains. In the drawing below, the red molecule is in an electronic excited and vibrational state. In a collision, it transfers some of its vibrational energy to the blue molecule.
Radiationless Transitions: Internal Conversion
If electrons can get to a lower energy state, and give off a little energy at a time, by hopping down to lower and lower vibrational levels, do they need to give off a giant photon at all? Maybe they can relax all the way down to the ground state via vibrational relaxation. That is certainly the case. Given lots of vibrational energy levels, and an excited state that is low enough in energy so that some of its lower vibrational levels overlap with some of the higher vibrational levels of the ground state, the electron can hop over from one state to the other, without releasing a photon.
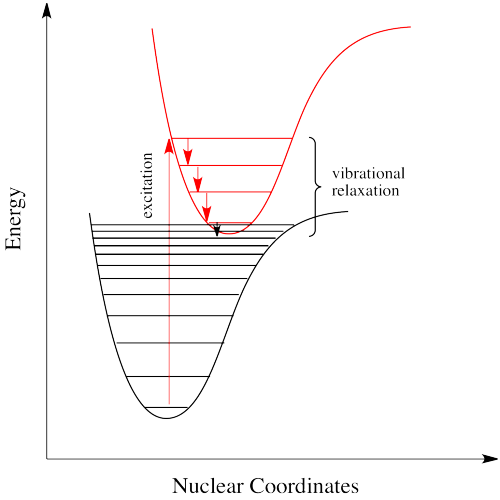
This event is called a "radiationless transition", because it occurs without release of a photon. The electron simply slides over from a low vibrational state of the excited electronic state to a high vibrational state of the electronic ground state. We will see a couple of iinds of radiationless transitions. Specifically, if the electron simply keeps dropping a vibrational level at a time back to the ground state, the process is called "internal conversion".
Internal conversion has an important consequence. Because the absorption of UV and visible light can result in energy transfer into vibrational states, much of the energy that is absorbed from these sources is converted into heat. That can be a good thing if you happen to be a marine iguana trying to warm up in the sun after a plunge in the icy Pacific. It can also be a tricky thing if you are a process chemist trying to scale up a photochemical reaction for commercial production of a pharmaceutical, because you have to make sure the system has adequate cooling available.
Radiationless Transitions: Intersystem Crossing
There is a very similar event, called "intersystem crossing", that leads to the electron getting caught between the excited state and the ground state. Just as, little by little, vibrational relaxation can lead the electron back onto the ground state energy surface, it can also lead the electron into states that are intermediate in energy.
For example, suppose an organic molecule undergoes electronic excitation. Generally, organic molecules have no unpaired electrons. Their ground states are singlet states. According to one of our selection rules for electronic excitation, the excited state must also have no unpaired electrons. In other words, the spin on the electron that gets excited is the same after excitation as it was before excitation.
However, that's not the lowest possible energy state for that electron. When we think about atomic orbital filling, there is a rule that governs the spin on the electrons in degenerate orbitals: in the lowest energy state, spin is maximized. In other words, when we draw a picture of the valence electron configuration of nitrogen, we show nitrogen's three p electrons each in its own orbital, with their spins parallel.

The picture with three unpaired electrons, all with parallel spins, shows a nitrogen in the quartet spin state. Having one of those spins point the other way would result in a different spin state. One pair of electrons in the p level would be spin-paired, one up and one down, even though they are in different p orbitals. That would leave one electron without an opposite partner. The nitrogen would be in a doublet spin state.
That isn't what happens. The quartet spin state is lower in energy than the doublet state. That's just one of the rules of quantum mechanics: maximize spin when orbitals are singly occupied.
It's the same in a molecule. The triplet state is lower in energy than the singlet state. Why didn't the electron get excited to the triplet state in the first place? That's against the rules. But sliding down vibrationally onto the triplet state from the singlet excited state isn't, because it doesn't involve absorption of a photon.
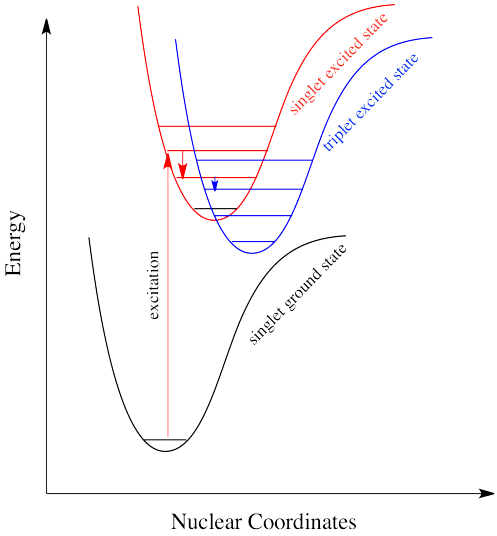
Intersystem crossing can have important consequences in reaction chemistry because it allows access to triplet states that are not normally avaiable in many molecules. Because triplet states feature unpaired electrons, their reactivity is often typified by radical processes. That means an added suite of reactions can be accessed via this process.
Phosphorescence: A Radiationless Transition Followed by Emission
Intersystem crossing is one way a system can end up in a triplet excited state. Even though this state is lower in energy than a singlet excited state, it can't be accessed directly via electronic excitation because that would violate the spin selection rule.
That's where the electron gets stuck, though. The quick way back down to the bottom is by emitting a photon, but because that would involve a change in spin state, it isn't allowed. Realistically speaking, that means it takes a long time. By "a long time", we might mean a few seconds, several minutes, or possibly even hours. Eventually, the electron can drop back down, accompanied by the emission of a photon. This situation is called "phosphorescence".
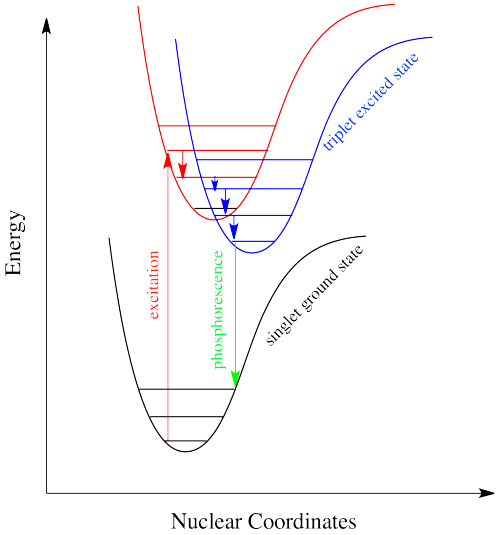
Molecules that display phosphorescence are often incorporated into toys and shirts so that they will glow in the dark.
Photosensitization
We have already seen that an excited state molecule can transfer some vibrational energy to another molecule via a collision. What about the energy of the electroic excited state? Can a molecule transfer a large quantum of energy to another -- essentially a photon's worth, but without the photon? The answer is yes.
In a collision, one molecule in an electronic excited state can transfer its energy to another. In the process, the first molecule returns to the ground state and the second is excited. This process is called "photosensitization".
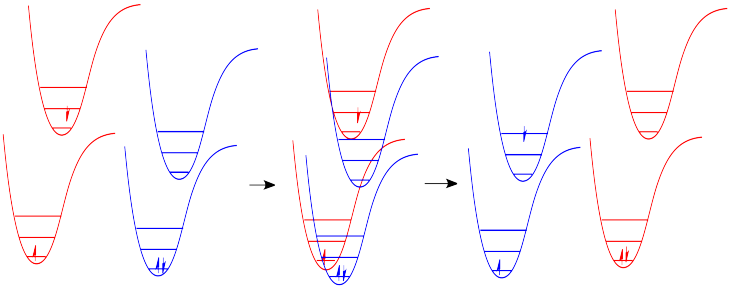
Photosensitization can occur in a couple of different ways. Because photosensitization does not involve absorption or emission of a photon, it can also lead to formation of a triplet excited state.
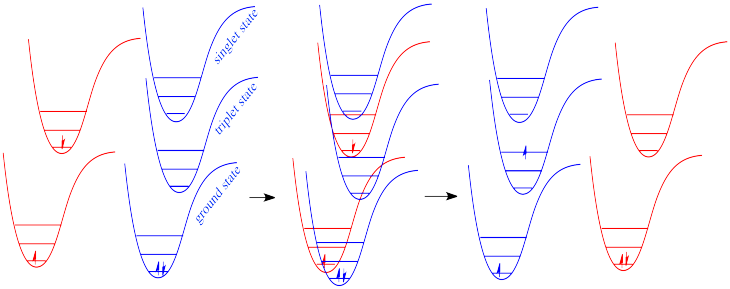
The significance of photosensitization is that compounds that do not have strong chromophores can still access electronic excited states if they come into contact with other molecules that do have strong chromophores. There are a number of compounds that are routinely used to induce excitation in other molecules; these photochemical enablers are referred to as photosensitizers.
Exercise \(\PageIndex{1}\)
Photoredox processes have experienced a great deal of attention from researchers. (For a nice overview, see Stephenson, J. Org. Chem. 2012, 77, 1617-1622).
- Photochemically excited species are often good reducing agents (or photoredox agents). Show why with the help of a generic energy diagram showing a HOMO → LUMO excitation.
- Ru(II) complexes such as Ru(bpy)32+ are good photoredox agents. Comment on the usefulness of Ru(bpy)32+ as a regular reducing agent (in its ground state), based on the following equation.
\[\ce{Ru(bpy)3^{3+} + e^{-} -> Ru(bpy)3^{2+}}\) \(E^{0} = 1.26V \nonumber\]
c) Calculate the energy difference between states suggested by this standard reduction potential.
d) Ru(bpy)32+ absorbs very strongly in the visible spectrum (450 nm). What colour is it?
e) What type of transition do you think this is?
f) Calculate the energy difference from the ground state to the initially formed excited state Ru(bpy)32+*.
g) What is the multiplicity of this initially formed excited state (e.g. a singlet or a triplet etc)? Show why.
h) The initially formed excited state Ru(bpy)32+* undergoes internal conversion (radiationless relaxation) and intersystem crossing to form triplet Ru(bpy)32+* before phosphorescence occurs at 615 nm. What colour is observed upon phosphorescence?
i) Calculate the energy difference from the triplet Ru(bpy)32+* to the ground state.
j) The difference between the wavelength absorbed and the wavelength emitted during fluorescence or phosphorescence is called the Stokes shift. What is the Stokes shift in this case?
k) Calculate the amount of energy transferred to vibrational states during the internal conversion.
l) Calculate the potential for the following reaction:
\(\ce{Ru(bpy)3^{3+} + e^{-} -> Ru(bpy)3^{2+*}}\) (triplet state) \(E^{0} = ?V\)
m) Compare the usefulness of Ru(bpy)32+ +vs. Ru(bpy)32+*as a reducing agent.
- Answer b
-
b) Ru(bpy)32+ would be a terrible reducing agent. The reduction potential of Ru(bpy)33+ is very high. That means Ru(bpy)32+ would not give up an electron very easily.
- Answer c
-
c) ΔG = - n F E0
ΔG = - 1 x 97,485 J V-1 mol-1 x 1.26 V = -123, 000 J mol-1 = -123 kJ mol-1
- Answer d
-
d) It absorbs in the blue part of the spectrum and appears orange.
- Answer e
-
e) It absorbs very strongly, so probably not d-d. It is probably MLCT, from the ruthenium to the π* in the bpy ligand.
- Answer f
-
f) E = h c / λ
E = (6.625 x 10-34 J s mol-1 x 3.0 x 108 m s-1 )/ (450 nm x 10-9 m nm-1) = 4.42 x 10-19 J
That's for one molecule. On a per mole basis, E = 4.42 x 10-19 J x 6.02 x 1023 mol-1 = 266,000 J mol-1 = 266 kJ mol-1.
- Answer g
-
g) Ru2+ would be low spin d6, a singlet ground state. The excited state will also be a singlet state.
- Answer h
-
h) 615 nm is in the orange region of the spectrum. This is light given off by the complex as it relaxes, so it is the colour we see.
- Answer i
-
i) E = h c / λ
E = (6.625 x 10-34 J s mol-1 x 3.0 x 108 m s-1 )/ (615 nm x 10-9 m nm-1) = 3.23 x 10-19 J
That's for one molecule. On a per mole basis, E = 3.23 x 10-19 J x 6.02 x 1023 mol-1 = 195,000 J mol-1 = 195 kJ mol-1.
- Answer j
-
j) Stokes shift = 615 nm - 450 nm = 165 nm
- Answer k
-
k) ΔE = E1 - E2 = 266 - 195 kJ mol-1 = 71 kJ mol-1
However, it would not be lost all at once, but in small increments equivalent to the differences between vibrational states.
- Answer l
-
l) If we were to reduce Ru(bpy)33+ directly into an excited state, we would arrive at a state much higher in energy than the ground state. That reduction would be harder to accomplish. In this case, the ending state would be 195 kJ mol-1 higher than Ru(bpy)32+.
ΔG = 195 - 123 kJ mol-1 = 72 kJ mol-1
E0 = - ΔG / n F = - (72 kJ mol-1 x 1000 kJ J-1) / (1 x 96,485 J V-1 mol-1) = -0.75 V
- Answer m
-
m) The oxidation of Ru(bpy)32+* would be very favourable compared to the oxidation of Ru(bpy)32+. The former is a much better reducing agent.