
9.2: Transformations in the TCA Cycle
- Page ID
- 189975

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\dsum}{\displaystyle\sum\limits} \)
\( \newcommand{\dint}{\displaystyle\int\limits} \)
\( \newcommand{\dlim}{\displaystyle\lim\limits} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\(\newcommand{\longvect}{\overrightarrow}\)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)In this section, we will take a look at the individual steps that make up the TCA cycle. We will try to see how the molecules are changing, and relate those changes to standard events from organic reactivity.
The very first step that leads to the citric acid cycle is the the conversion of pyruvate to acetyl coenzyme A (or acetyl CoA).

Acetyl coenzyme A is a very important molecule in a wide range of biochemical reactions, in addition to its role in the citric acid cycle.
So, this is a really important transformation, yet this step poses a vexing problem. If you look at the reaction, you will see that one of the original three carbon atoms in pyruvate is lost as carbon dioxide. This is called a decarboxylation, and it is not uncommon in biological chemistry. A decarboxylation starts with a carboxylate or carboxylic acid group, which already contains a C=O bond as well as a C-O bond. A lone pair from the C-O oxygen donates to the carboxyl carbon, making the second C=O bond. That step displaces a leaving group from the other side of the carboxyl carbon. Most commonly, the leaving group needed for this step is a stable, enolate anion.
At first glance, things might seem to be OK for that to proceed in the pyruvate decomposition step, but there is a problem. The leaving group is not an enolate anion, and it isn't a particularly stable anion. The most stable carbon anions are resonance-delocalised, preferably over oxygen, as in an enolate or carbonate anion. The leaving group that we would have here, however, offers no such advantage.

Exercise \(\PageIndex{1}\)
Show, with an additional resonance structure, why resonance delocalisation of the acyl anion is not possible.
- Answer
-

Exercise \(\PageIndex{2}\)
Show, using molecular orbital cartoons, why resonance delocalisation of the acyl anion is not possible.
- Answer
-

Something else is also happening in the pyruvate-to-acetyl CoA step. A thiolate group is also incorporated at this stage; in fact, this part brings in the coenzyme A. From that point of view, maybe this step is best thought of as a carboxylic substitution at the acyl group. Thiols can certainly displace leaving groups from carboxyloids.
Unfortunately, we hit the same roadblock this way. We still need a carbonyl-based anion, and that isn't going to happen. Either way, the enzyme-catalysed strategy for this step must address stabilisation of that carbonyl fragment as an anion. That task is going to be accomplished through temporary attachment to an ylide, which we will see in the next section.
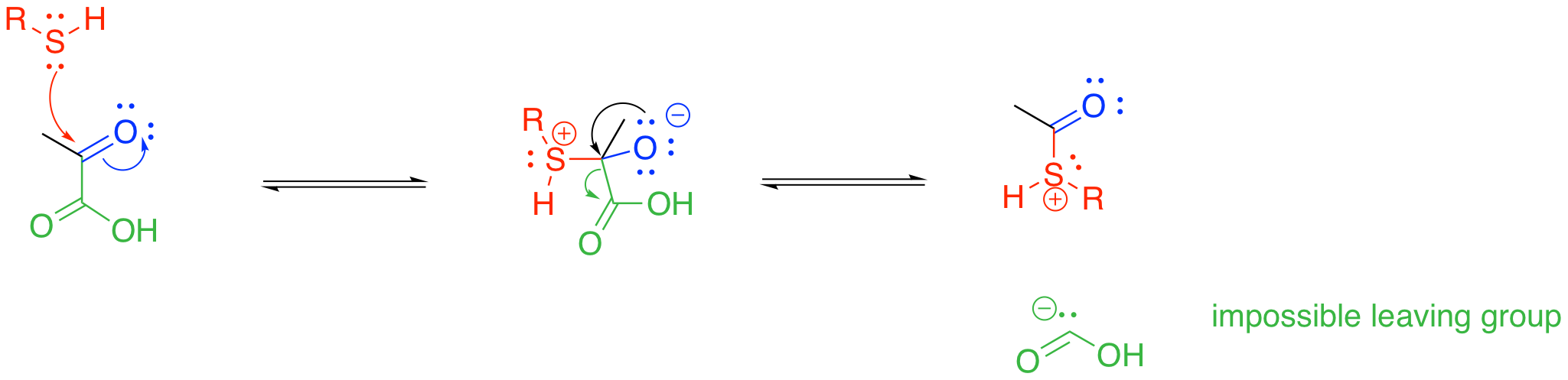
The TCA cycle really gets started with the addition of the acetyl CoA to oxaloacetate, forming citric acid. That step also results in displacement of the CoA thiol via hydrolysis.

This citrate-forming step is a little easier to work through than that last one that we saw. The key is to recognise that the acetyl group, which contains a carbonyl and an alpha position, could easily tautomerize to form an enol. Viewed that way, the acetyl part of the acetyl CoA as a nucleophile, rather than an electrophile.

The best electrophile in the complementary molecule, the oxaloacetate, is the carbonyl of the ketone; remember, pi-donation, diminishes the electrophilicity of the carboxylate groups, to say nothing of the negative charge. So what we have here is a standard addition of a nucleophile to a carbonyl, with the latter forming a hydroxy group as it picks up a proton. With an enol or enolate nucleophile, it's an aldol reaction.
Furthermore, the displacement of the thiol by water seems pretty trivial. That's standard carboxyloid substitution.
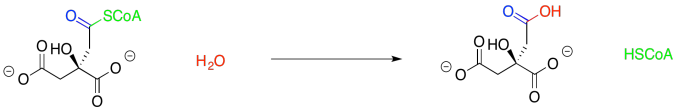
The subsequent conversion of citrate to isocitrate appears, at first, to be a little trickier. At one position, a hydroxy group is replaced by a hydrogen atom, whereas a hydroxy group replaces a hydrogen atom at another position.

This one isn't terribly complicated either, though. It helps to see that the reaction is accomplished in two separate, and completely complementary, steps. You have seen the first one before in connection with aldol reactions. Sometimes an aldol reaction is followed by loss of a water molecule. The newly-made OH group picks up a proton from the neighbouring alpha position to form that water. Remember, the older term, "aldol condensation", refered to the formation of water, which would condense on the glassware in which the reaction occurred. This step is often promoted by the presence of additional conjugation involving the new double bond and sometimes it is promoted by heat, but in this case it is carried out by an anzyme.
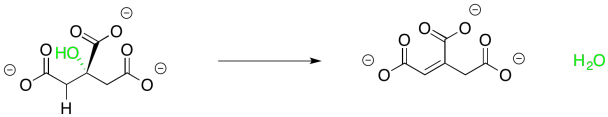
Note that we rotated around that middle bond of citrate, on the left, but it's still the same molecule.
We may not have considered the reversibility of this reaction before, but that's what we need to do now. The enone group, a carbonyl in conjugation with a C=C bond, is electrophilic at both the carbonyl carbon and the beta carbon. The water molecule could simply add back in where it used to be, making citrate again. There is another way to look at the electrophile, however, because there is another carbonyl at the other end of the double bond. Adding in that direction leads to isocitrate.
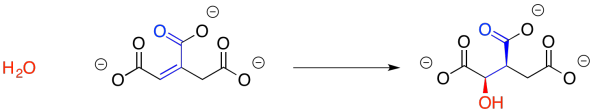
Exercise \(\PageIndex{3}\)
Draw resonance structures that demonstrate the electrophilicity of the conjugated positions in the intermediate above.
- Answer
-
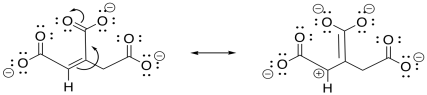
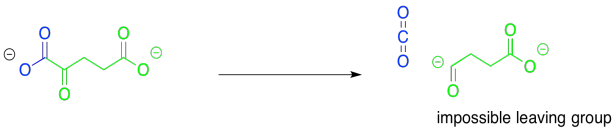
During the TCA cycle, two additional carbons are lost as carbon dioxide, as the six-carbon citrate makes its way back to the four-carbon oxaloacetate. The first one is lost in the conversion of isocitrate to alpha-ketoglutarate. Also in this step, a hydroxy group is converted into a carbonyl.

So, this step is another case of decarboxylation. We saw that event in the lead-in to the TCA cycle, formation of acetyl CoA. Just as we saw in that case, we need a stable leaving group for the carbon dioxide to leave behind, but it doesn't look like we have that here, either. Remember, it is best to have a resonance-stabilised carbon anion, but we don't have one.

This time, the other half of the reaction could be very helpful. If that alcohol is coverted to a ketone, the problem of decarboxylation is solved because it would leave behind an enolate anion. The enolate anion is resonance-stabilised. That situation would be ideal for decarboxylation.
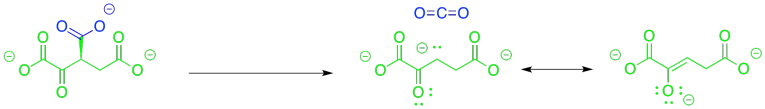
From the point of view of the kinds of reactions with which you are most familiar at this point, the more difficult part about this step is getting from the alcohol group to the ketone. That's a fairly common transformation, though, and in biology it is often accomplished with the common cofactor, NAD+.

Why do we need this cofactor? If you analyse the changes in the atoms from one structure to the next, you will notice that the conversion of the alcohol to a ketone requires the loss of two hydrogen atoms. A beginning student will often notice that fact and remove the protons in the best way that they know: as protons.

That will not work. If we just lose two protons, then we build up an additional 2- charge on the molecule. Electrons are very small, but they are very important, and they don't just disappear because it would make things more convenient. Now, there is a class of biological cofactors that will take care of this problem. These cofactors are biochemical oxidants, and NAD+ is a member. It can accept two, and only two, electrons at a time. And that's exactly what we need.
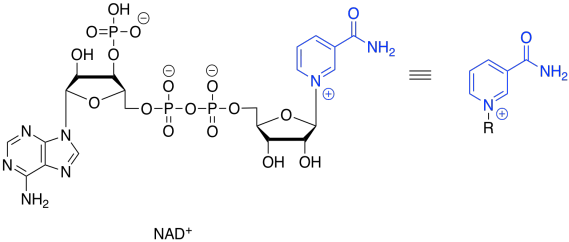
NAD+ can accept a pair of electrons because it is very electrophilic. That's partly because of the full positive charge. It also contains an electron-withdrawing carbonyl. These two factors combine to make this cofactor a good electrophile.
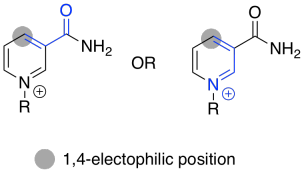
Exercise \(\PageIndex{4}\)
Use resonance structures to confirm the electrophilicity of the indicated position.
When NAD+ accepts this electron pair, it becomes NADH, a biochemical reductant. It's worth looking at that for a moment because you have seen the sort of reaction that NADH can accomplish: the delivery of a hydride nucleophile (H-) to a carbonyl electrophile. That would convert a ketone to an alcohol, for example. That's exactly the opposite of what we are doing here. Oxidation of the alcohol by NAD+ is what's called the "microscopic reverse" of the hydride addition, or reduction, of the ketone to the alcohol. The reaction follows the same pathway, but in the opposite direction. Instead of NADH pushing a hydride ion onto an electrophilic carbonyl, the alcohol is pushing a hydride ion onto the electrophilic NAD+.
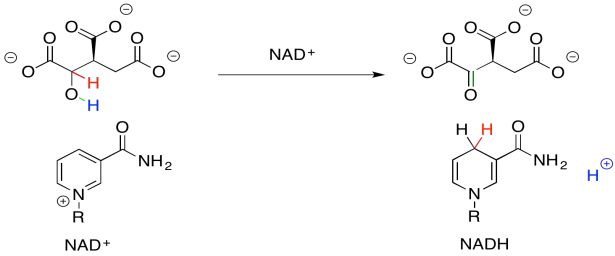
The very next step of the TCA cycle is also a decarboxylation step.

Once again, the major consideration here is the presence of a good leaving group. This time, the reaction looks an awful lot like that original step, the conversion of pyruvate to acetyl CoA. Decarboxylation will provide an anion that sits directly on the carbonyl carbon, rather than alpha to it. In fact, this problem will be solved in exactly the same way as in that first step. The pathway will involve temporary attachment of an ylide to stabilise the anion.
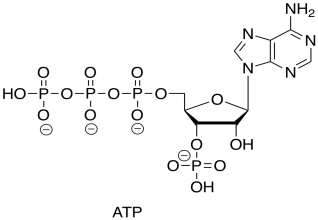
We now come to the ATP-producing step in the TCA cycle. Actually, ATP is not produced directly, but is coupled to a GTP-GDP cycle.

You recall that ATP is the triphosphate ester of the nucleotide, adenosine.
GDP is just the diphosphate ester of another nucleotide, guanosine.
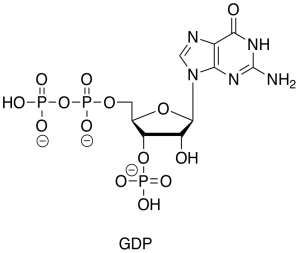
At first glance, it looks like the GDP is just picking up another phosphate group from inorganic phosphate ion (Pi is a biochemistry shorthand for phosphate ion, whether it is present as PO43-, HPO42-, or H2PO4-). For its part, the succinyl coenzyme A looks like it is simply getting hydrolysed: reacting directly with water to displace the thiol leaving group.
In reality, the succinyl CoA acts as a primer for the phosphate group, activating it as a more electrophilic phosphoanhydride. That step makes it easier for the GDP to donate to it as a nucleophile, leading to an additional phosphate connection.

The GDP simply displaces a carboxylate leaving group from the phosphoanhydride.

At this point, we have accomplished a lot in terms of the overall changes in the TCA cycle. We picked up a two-carbon piece in the form of an acyl group (CH3CO). We have made a molecule of ATP. We have unloaded those two extra carbons as carbon dioxide and are back at a four-carbon diacid again; we have almost regenerated the oxaloacetate. We still have a couple of things to do in order to complete the cycle. We need to install another carbonyl in the four-carbon chain; that is, we need to add in an oxygen, somehow. On the way, we are going to produce some more NADH and FADH2, which are also important in meeting the cell's energy needs, as well as its synthetic feedstock requirements.
To that end, succinate is first converted to fumarate. That's going to give the molecule an electrophilic position where it is needed, to allow the installation of that oxygen atom.

If you think about it, this step is similar to one you have seen before on this page. We need to make a double bond where there was previously a single bond. In order to do that, we need to lose two hydrogen atoms.

Losing two protons seems simple enough; that's especially true in this case, because each proton would be lost from an alpha position. Those alpha positions are ever-so-slightly acidic. However, that doesn't lead all the way to malate, because we still have two extra electrons to deal with.
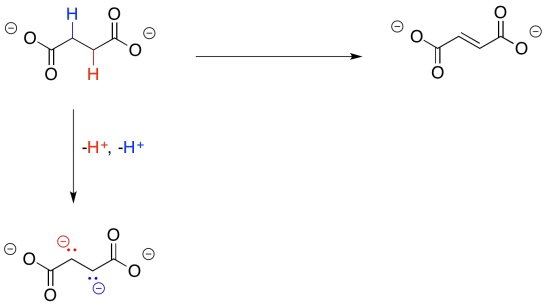
Again, we dealt with this problem earlier. All we needed was an oxidant, which would accept the two extra electrons. This time, instead of NAD+, the process will make use of a molecule of FAD.
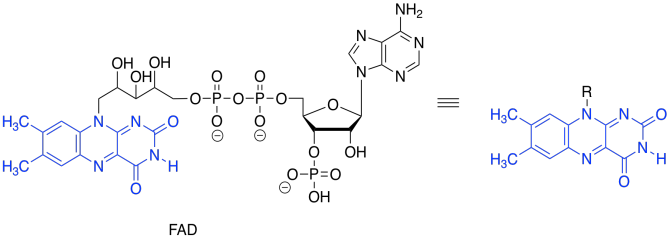
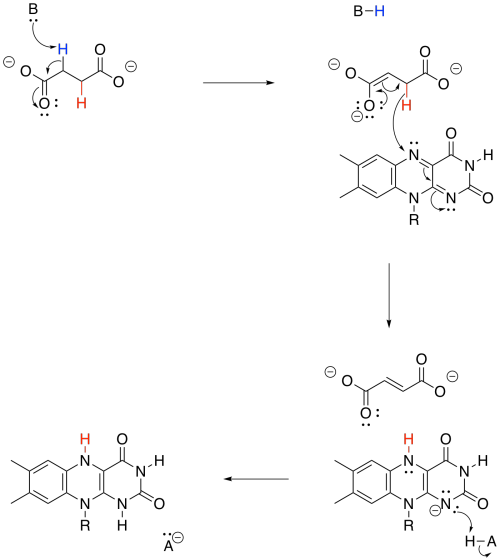
Just like the conversion of NAD+ to NADH, the transformation of FAD to FADH2 can absorb a hydride ion. That's a proton plus a lone pair.
Exercise \(\PageIndex{5}\)
Overall, FAD takes up both a hydride (H-) and a proton (H+) in this step. We think of the hydride as adding to the nitrogen atom next to the benzene. Use resonance structures to show why that is probably the case.
We are almost there. Now, an oxygen atom is installed in the form of an OH group.

Of course, at this point it seems pretty clear that that hydroxy group just comes from a 1,4-addition of water to the enone electrophile of the fumarate.

Finally, the resulting malate is converted back into oxaloacetate, completing the cycle. You have seen steps very similar to this one, earlier in the cycle. At this point we are ready to start the cycle again.

Exercise \(\PageIndex{6}\)
Before reading further, explain the role of the NAD+ in the final step of the TCA cycle.
Exercise \(\PageIndex{7}\)
The malate and oxaloacetate molecules in the above drawing are oriented in two different directions. Rotate the malate so that it more closely corresponds with the structure of the oxaloacetate.
So, if you couldn't remember where you have seen this sort of change before, take a closer look. The conversion of malate to oxaloacetate involves the loss of two hydrogen atoms. That's two protons plus two electrons.

We can make this happen if we have an electron acceptor. A biochemical oxidant, such as NAD+, is a great candidate. It will accept a hydride ion from the malate, helping it on its way to oxaloacetate.
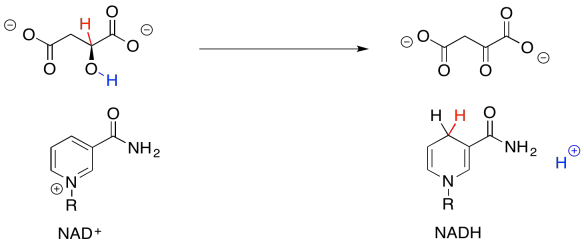
In the next section, we will take a closer look at the mechanisms that these compounds follow as they proceed through the TCA cycle.