
20.14: Primary Protein Structure
- Page ID
- 49623

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)Proteins which occur in nature differ from each other primarily because their side chains are different. Partly this is a matter of composition. In wool, for example, 11 percent of the side chains are cysteine, while no cysteine occurs in silk at all. To a much larger extent though, the differences between different proteins is a matter of the sequence in which the different side chains occur. This is especially true of globular proteins like enzymes. The sequence of side chains along the backbone of peptide bonds in a polypeptide is said to constitute its primary structure.
The 20 different amino acids permit construction of a tremendous variety of primary structures. Consider, for example, how many tripeptides similar to that shown in Eq. 1 on the page on polypeptide chains can be constructed from the 20 amino acids. In the example shown, the first amino acid in the chain is glycine, but it might just as well be proline or any other of the 20 amino acids. Thus there are 20 possibilities for the first place in the chain. Similarly there are 20 possibilities for the second place in the chain, making a total of 20 × 20 = 400 possible combinations. For each of these 400 structures we can again choose from among 20 amino acids for the third place in the chain, giving a grand total of 400 × 20 = 203 = 8000 possible structures for the tripeptide.
A general formula for the number of primary structures for a polypeptide containing n amino acid units is 20n—a very large number indeed when you consider that most proteins contain at least 50 amino acid residues. [2050 = (2 × 10)50 = 250 × 1050 = 1015 × 1050 = 1065] Primary structure is conventionally specified by writing the three-letter abbreviations for each amino acid, starting with the —NH3+ end of the polymer. In some cases, this is even shortened to the 1-letter abbreviation sequence. For example, the structure
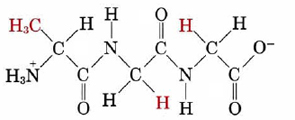
which reading from the -NH3+ end is alanine, glycine, glycine would be specified as
Ala-Gly-Gly or AGG
Note that Ala-Gly-Gly is not the same as Gly-Gly-Ala. In the latter case glycine rather than alanine has the free —NH3+ group. Because its ends are different, there is a directional character in the polypeptide chain.
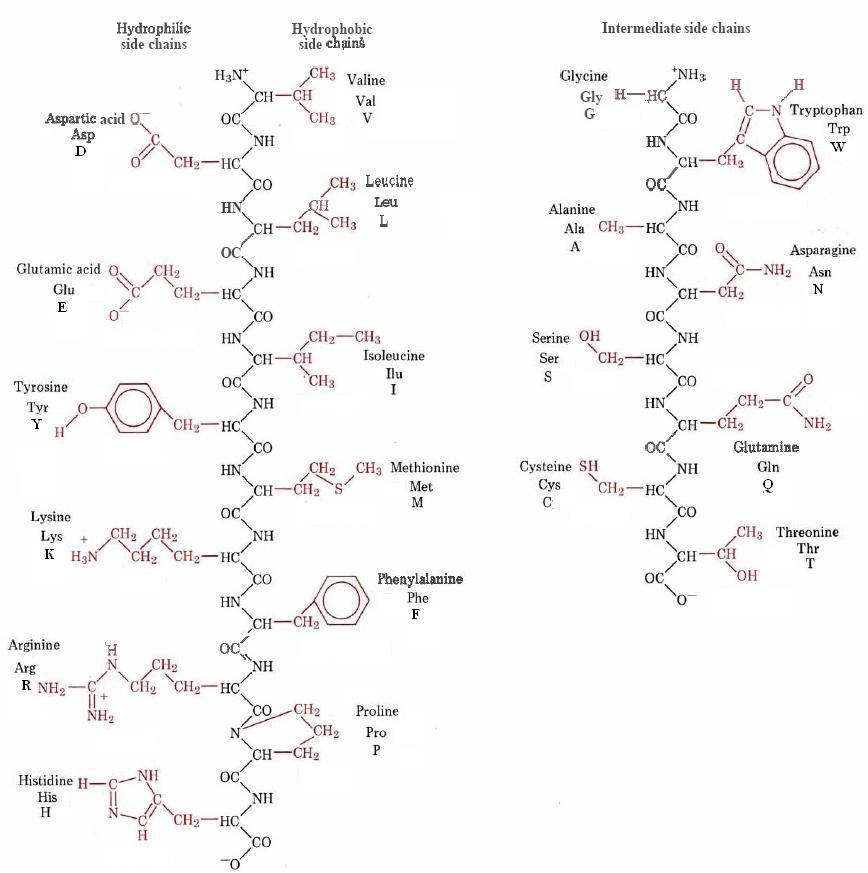
Both three letter and single letter abbreviations are shown in Figure \(\PageIndex{1}\) :
Determination of the primary structure of a protein is a difficult and complicated problem. It also is a rather important one—the sequence of amino acids governs the three-dimensional shape and ultimately the biological function of the protein. Consequently much effort has gone into methods by which primary structure can be elucidated.
Insulin was the first protein whose amino acid sequence was determined. This pioneering work, completed in 1953 after some 10 years of effort, earned a Nobel Prize for British biochemist Frederick Sanger (born 1918). He found the primary structure to be
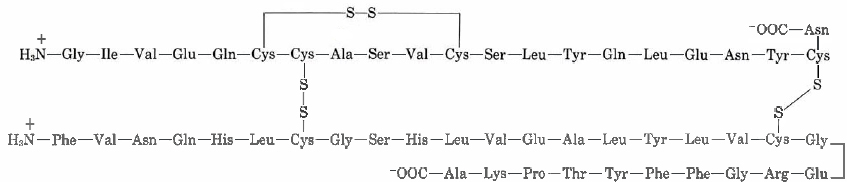
Note how there are two chains in this structure, one with 21 side chains and the other with 30. These two chains are linked in two places with disulfide (—S—S—) bridges, each connecting two cysteine residues in different chains.
In order to determine the primary structure of a protein, a known mass of pure sample is first boiled in acid or base until it is completely hydrolyzed to individual amino acids. The amino acid mixture is then separated chromatographically and the exact amount of each amino acid determined. In this way, one can find that for every 3 mol serine in the insulin molecule, there are 6 mol leucine. The next step is to break down the protein into smaller fragments. Disulfide bridges are broken by oxidation after which the protein is selectively hydrolyzed by enzymes, called proteases, such as trypsin or chymotrypsin. In a favorable case one will then have several fragments each containing 10 or 20 amino acid residues. These can then be separated and analyzed individually.
Using Edman degradation, so named for Pehr Edman, the sequence of amino acids in one of these polypeptide fragments is usually determined using phenylisothiocyanate, 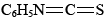 , which selectively attacks the —NH3+ end of the polypeptide chain. This reaction is carried out under basic conditions. Addition of acid then splits off the terminal amino acid, and it can be identified. Since the rest of the polypeptide chain is left intact, this process can be repeated, and each amino acid in the sequence can be attacked, removed, and identified. By snipping off amino acids one at a time, one eventually finds the complete sequence for the fragment. This whole process can be automated and hence sped up considerably. Once the fragments have been sequenced, it becomes a matter of ordering them correctly. Since different proteases hydrolyze peptide bonds at different places in the amino acid sequence, different fragmentation patterns can be used to determine the sequence for the whole protein. The end of a fragment from a trypsin digest will be in the middle of a fragment from a chymotrypsin digest, for instance. This provides a relatively quick means to sequence unknown protein sequences.
, which selectively attacks the —NH3+ end of the polypeptide chain. This reaction is carried out under basic conditions. Addition of acid then splits off the terminal amino acid, and it can be identified. Since the rest of the polypeptide chain is left intact, this process can be repeated, and each amino acid in the sequence can be attacked, removed, and identified. By snipping off amino acids one at a time, one eventually finds the complete sequence for the fragment. This whole process can be automated and hence sped up considerably. Once the fragments have been sequenced, it becomes a matter of ordering them correctly. Since different proteases hydrolyze peptide bonds at different places in the amino acid sequence, different fragmentation patterns can be used to determine the sequence for the whole protein. The end of a fragment from a trypsin digest will be in the middle of a fragment from a chymotrypsin digest, for instance. This provides a relatively quick means to sequence unknown protein sequences.
Edman degradation is not the only method for which proteins are sequenced nowadays. Mass spectrometry can sequence polypeptides of 20 to 30 amino acids in length, using a technique called tandem mass spectrometry. In this method, a polypeptide is sent through one mass spectrometer, which ionizes the polypeptide. The charged peptide then enters a collision chamber, causing the peptide to fragment at different peptide bonds. The resulting fragments are then measured by a second mass spectrometer. The resultant spectrum can determine the peptide sequence by differences in mass of the fragments.
With the advent of DNA sequencing methods and the success of the Human Genome Project, many protein sequences are now determined indirectly, through the genetic code. When the DNA sequence for a protein is known, this can be used to determine the protein sequence. By the same token, a known protein sequence can be used to determine the gene coding for that protein. Thus there are many different ways to determine protein sequences, all of which compliment each other.[1]
As methods for determining primary structure have become more advanced, a great many proteins have been sequenced, and some interesting comparisons can be made. A particularly intriguing example is that of cytochrome c, an electron carrier which is found in all organisms that use oxygen for respiration. When samples of cytochrome c from different organisms are compared, it is found that the amino acid sequence is usually different in each case. Moreover, the more widely separated two species are in their macroscopic features, the greater the degree of difference in their protein sequences. When horse cytochrome c is compared with that of yeast, 45 out of 104 residues are different. Only two substitutions are found between chicken and duck, and cytochrome c is identical in the pig, cow, and sheep. The magnitudes of these changes coincide quite well with biological taxonomy based on macroscopically observable differences. Cytochrome c can be used to trace biological evolution from unicellular organisms to today’s diverse species, and even to estimate times at which branching occurred in the family tree of life. This makes molecular methods a powerful tool for the evolutionary biologist as well.
References
- ↑ Nelson, D.L., Cox, M.M. Lehninger Principles of Biochemistry(5th ed). New York: W.H. Freeman and Company, 2008. pp. 96-100.