
1.9: Crystallographic computing
- Page ID
- 352428

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\dsum}{\displaystyle\sum\limits} \)
\( \newcommand{\dint}{\displaystyle\int\limits} \)
\( \newcommand{\dlim}{\displaystyle\lim\limits} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\(\newcommand{\longvect}{\overrightarrow}\)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)Readers who have arrived at this chapter in a sequential manner will notice that, apart from the phase problem, the relationship between the diffraction pattern (reciprocal space) and the crystal structure (direct space) is mediated by a Fourier transform represented by the electron density function: \(ρ(xyz)\) (see the drawing on the left).
Readers will also know that the relationship between these two spaces is "holistic", meaning that the value of this function, at each point in the unit cell of coordinates \((xyz)\), is the result of "adding" the contribution of "all" structure factors [ie diffracted waves in terms of their amplitudes \(|F(hkl)|\) and phases \(Φ(hkl)\)] contained in the diffraction pattern. They will also remember that the diffraction pattern contains many structural factors (several thousand for a simple structure, and hundreds of thousands for a protein structure).
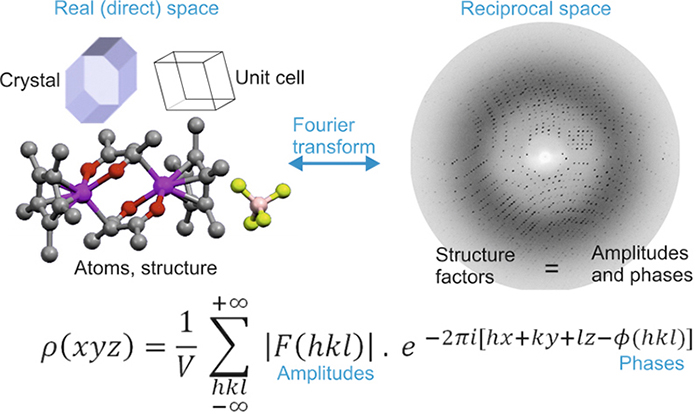
The "jump" between direct and reciprocal spaces, mediated by a Fourier transform represented by the electron density function
Moreover, the number of points in the unit cell, where the ρ function has to be calculated, is very high. In a cell of about 100 x 100 x 100 Angstrom3, it would be necessary to calculate at least 1000 points in every unit cell direction to obtain a resolution of 100/1000, which equals 0.1 Angstrom in each direction. This means calculating at least 1000 x 1000 x 1000 = 1,000,000,000 points (one billion points) and at each point to "add" several thousand (or hundreds of thousands) structure factors F(hkl).
It should therefore be clear that, regardless of the difficulties of the phase problem, solving a crystal structure implies the use of computers.
Finally, the analysis of a crystal or molecular structure also implies calculating many geometric parameters that define interatomic distances, bond angles, torsional angles, molecular surfaces, etc., using the atomic coordinates (xyz).
The "hardware"
For the reasons described above, since the beginning of the use of Crystallography as a discipline to determine molecular and crystal structures, crystallographers have devoted special attention to the development of calculation tools to facilitate crystallographic work. With this aim, and even before the early computers appeared, the crystallographers introduced the so-called "Beevers-Lipson strips," which were widely used in all Crystallography laboratories.


The Beevers-Lipson strips
The Beevers-Lipson strips (which were strips of paper containing the values for some trigonometric functions) were used in laboratories to speed up the calculations (by hand) of the Fourier transforms (see above: the electron density function, for example).
These strips were introduced in 1936 by A.H. Beevers and H. Lipson. In the 1960s, more than 300 boxes were distributed to nearly all the laboratories in the world. You can also have a look into the description made by the International Union of Crystallography. The nightmare was maintaining upright this box, which had a very narrow base, otherwise it was impossible to maintain the strips correctly stored!
As expected, the introduction of early computers (or electro-mechanic calculators) inspired great hope in crystallographers...


ENIAC (Electronic Numerical Integrator and Computer, 1945) -- the very first electronic computer. Some pictures of the rooms where it was installed.
ENIAC, short for Electronic Numerical Integrator And Computer, was the first general-purpose electronic computer, whose design and construction were financed by the United States Army during the Second World War. It was the first digital computer capable of being reprogrammed to solve a full range of computing problems, especially calculating artillery firing tables for the U.S. Army's Ballistic Research Laboratory.
The ENIAC had immediate importance. When it was announced in 1946, it was heralded in the press as a "Giant Brain". It boasted speeds one thousand times faster than electro-mechanical machines, a leap in computing power that no single machine has matched. This mathematical power, coupled with general-purpose programmability, excited scientists and industrialists.
Besides its speed, the most remarkable thing about ENIAC was its size and complexity. ENIAC had 17,468 vacuum tubes, 7,200 crystal diodes, 1,500 relays, 70,000 resistors, 10,000 capacitors and around 5 million hand-soldered joints. It weighed 27 tons, was roughly 2.6 m by 0.9 m by 26 m, took up 63 m², and consumed 150 kW of power.
Later, with the development of Electronics and Microelectronics, which introduced integrated circuits, computers became accessible to crystallographers, who flocked to these facilities with large boxes of "punched cards" (the only means for data storage at that time), containing the diffraction intensities and their own computer programs.

A punch card or punched card (or punchcard or Hollerith card or IBM card), is a piece of stiff paper which contains digital information represented by the presence or absence of holes in predefined positions. It was used by crystallographers until the end of the 1970s.


Punched paper tape (shown in yellow) and different magnetic tapes (as well as some small disks) used for data storage during the 1970s and 1980s.
Around the early 1970s, and for over a decade, crystallographers became a nightmare for the managers and operators of the so-called "computing centers,'' running in some universities and research centers.
In the 1980s the laboratories of Crystallography became "flooded" with computers, which for the first time gave crystallographers independence from the large computing centers. The VAX series of computers (sold by the company Digital Equipment Corporation) marked a splendid era for crystallographic calculations. They allowed the use of magnetic tapes and the first hard disk drives, with limited capacity (only a few hundred MB) -- very big and heavy, but they eliminated the need for the tedious punched cards. Nostalgics should have a look into this link.!!!

A typical computer (of the VAX series) used in many Crystallography laboratories during the 1980s.
Over the years, crystallographic computing has become easy and affordable thanks to personal computers (PC), which meet nearly all the needs of most conventional crystallographic calculations, at least concerning crystals of low and medium complexity (up to hundreds of atoms). Their relative low price and their ability to be assembled into "farms" (for distributed calculation) provide crystallographers the best solution for almost any type of calculation.


Left: A typical personal computer (PC) used in the 2000s
Right: A typical PC-farm used in the 2000s
However, the crystallography applied to macromolecules not only needs what we could call "hard" computing. The management of large electron density maps, which are used to build the molecular structure of proteins, as well as the subsequent structural analysis, requires more sophisticated computers with powerful graphic processors and, if possible, with the capability of displaying 3-dimensional images using specialized glasses...

A Silicon Graphics computer used to visualize 3-dimensional electron density maps and structures. The processor and the screen are complemented by an infrared transmitter (black box on the screen) and the glasses used by the crystallographer.
The current computing facilities represent a big jump respect to the capabilities available during the mid-twentieth century, as it is shown in the representation of the structural model used for the structural description of penicillin, based on three 2-dimensional electron density maps... And even 3d maps where also used!...


Left: Three-dimensional model of the structure of penicillin, based on the use of three 2-dimensional electron density maps, as used by Dorothy C. Hodgkin, Nobel laureate in 1964
Right: Representation of 3d electron density maps used until the middle of the 1970's. The contours are lines of electron density and show the positions of individual atoms in the structure
A typical personal computer commonly used since 2010 for crystallographic calculations and also for their graphic capabilities
The software
At present there are enough personal, institutional or commercial computer program developments, or even computing facilities through remote servers, to fulfill nearly all of the needs for crystallographic computing, as well as many sources from which one can download most of those programs. In this context, it could be useful to check the following links:
Crystallographic computer programs
- Macromolecules: The Web-Book of the Department of Crystallography & Structural Biology (CSIC)
- Of general interest: The crystallographic software list maintained by the International Union of Crystallography - (IUCr)
Specifically for compounds of small and medium size (molecular or not) we recommend using the Wingx package which can be freely downloaded by courtesy of Louis J. Farrugia, (University of Glasgow, UK). It is easy to install on a PC and contains an interface which includes the most important programs for small and medium size crystallographic problems. Also, for these types of compounds there is a very useful computer program (Mercury), user-friendly and free, which includes powerful graphics and some other analytical tools to analyze crystal structures. It can be downloaded from the Cambridge Crystallographic Data Centre, UK.
Protein crystallographers need more specific programs, and in this context we recommended using the link offered by CCP4, Collaborative Computational Project No. 4, Software for Macromolecular X-Ray Crystallography.
On the other hand, crystallographic work is currently unimaginable without having access to crystallographic databases, which contain all the structural information that is being published and which have a clear added value for the researcher. The type of structure is what determines its inclusion in any of the existing databases. Thus, metals and intermetallic compounds are made available in the database CRYSTMET; inorganic compounds are centralized in the ICSD database (Inorganic Crystal Structure Database); organic and organometallic in CSD (Cambridge Crystallographic Database); and proteins in PDB (Protein Data Bank), which is a databank (not a database). Other databases, databanks, etc., do not necessarily contain structural information in the most precise sense, but they can also be very helpful for crystallographers. And this is the case of WebCite published by the Cambridge Crystallographic Data Centre (CCDC), containing over 2000 articles with very important information for structural chemistry research in its broadest sense, and in particular to pharmaceutical drug discovery, materials design or drug development, among others.
Structural databases and databanks
- CRYSTMET: Metals and intermetallic compounds (license required)
- ICSD: Inorganic compounds (license required)
- CSD: Organic and organometallic compounds (license required)
- glycoSCIENCES.de: Carbohydrates
- LipidBank: Lipids
- PDB: Proteins, Nucleic acids and large complexes
- NDB: Nucleic acids
As indicated, some of these databases (or databanks) are public (glycoSCIENCES.de, LipidBank, PDB and NDB), and therefore can be searched online. However, others (CRYSTMET, ICSD and CSD) require a license or even a local installation.
During the period 1990-2012, CRYSTMET, ICSD and CSD have been licensed free of charge to all CSIC research institutes (CRYSTMET and ICSD) and to all academic institutions in Spain and Latin American countries (CSD). However, due to economic constraints, the CSIC's authorities decided to reduce drastically this program that was managed through the Department of Crystallography and Structural Biology (at the Institute of Physical Chemistry "Rocasolano"). Nowadays this program is maintained in a reduced manner, only for Spanish institutions, as it can be seen through this link.