
9: Multistep Synthesis (Experiment)
- Page ID
- 61319
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\dsum}{\displaystyle\sum\limits} \)
\( \newcommand{\dint}{\displaystyle\int\limits} \)
\( \newcommand{\dlim}{\displaystyle\lim\limits} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\(\newcommand{\longvect}{\overrightarrow}\)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)When synthesizing complex organic molecules, it is common to have at least a dozen individual transformations whereby the product of one reaction is then used as the starting material for the next reaction. You will have an opportunity to do a multistep synthesis starting with inexpensive, readily available benzaldehyde. The sequence you will attempt is first the conversion of benzaldehyde to benzoin using the vitamin, thiamin, as a catalyst. In the second step, the benzoin is oxidized to benzil through the use of an oxidizing agent. The third step is a condensation reaction of benzil with dibenzyl ketone (1,3-diphenyl-2-propanone) to produce tetraphenylcyclopentadienone. An alternative third step is the reduction of benzil to dihydrobenzoin with a reducing agent, sodium borohydride. An additional fourth step is possible converting the tetraphenylcyclopenta-dienone to a substituted naphthalene via a Diels-Alder reaction (followed by decarbonylation) using microwaves as the energy source.
One problem which becomes apparent is that the yield of the overall final product will be limited by the lowest yielding individual reaction. Therefore, each reaction in the sequence must be a high yielding reaction. Second, the overall final product yield is the product of each individual percentage yield. Therefore, if each step is a 90% yield and there are 10 steps, the overall final product yield is (0.90)10 or 35%. For a twenty step reaction, the overall yield would only be 12%. In a two step reaction, if one step had a yield of 50%, the highest overall yield possible is 50%. It is not necessary, or desirable, to use all of your material in each step.
Step 1: Synthesis of Benzoin
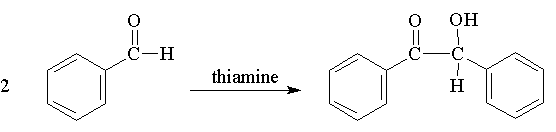
This reaction is a classic--the conversion of two molecules of an aldehyde to an alpha-hydroxy ketone. The reaction is known as a benzoin condensation ("condensation" because two molecules become condensed to one molecule). This reaction, which requires a catalyst, if often performed with cyanide ion. We will use thiamine as a catalyst. It is heat-sensitive and may decompose if heated too vigorously. Instead of running this reaction at elevated temperatures for a few hours, we will allow the reaction to proceed closer to room temperature for 24 hours or more. Benzaldehyde is easily oxidized to benzoic acid which can impede the desired reaction so freshly distilled benzaldehyde is used. The concentration of reactants and temperatures of solutions are critical to obtaining a good yield so procedures must be followed carefully. Too much water will force benzaldehyde out of solution preventing an efficient reaction. Too little water prevents the thiamine hydrochloride from dissolving. Some of the base reacts with the thiamine hydrochloride to produce thiamine which is the active catalyst.
Procedure: Place 1.5 mL of 5M NaOH (CAUTION: extremely caustic) in a 10 mL Erlenmeyer flask and cool in an ice bath. In a 50 mL Erlenmeyer flask dissolve 0.80 g of thiamine hydrochloride (MW=337) in 2.5 mL of water. Add 7.5 mL of 95% ethanol to the thiamine and cool the solution for several minutes in an ice bath. While keeping both flasks in the ice bath, add the 1.5 mL of previously cooled 5M sodium hydroxide dropwise (3-5 minutes) to the thiamine solution with swirling so that the solution stays below room temp. Remove the 50 mL flask from the ice bath, add 5.0 mL of benzaldehyde (d=1.044 g/mL) at one time, swirling the flask so that the benzaldehyde mixes with the yellow, aqueous, basic layer. The solution becomes milky but then clears*. Seal the flask with Parafilm and place it in your drawer until the next lab period.
*If the mixture does not go to solution (eg, if two layers are obvious), place the flask in a warm water bath at approx. 50 oC until the solution clears or for a maximum of 10 minutes. You can use hot water from the faucets at the front of the lab. The mixture should become homogeneous in the water bath but it may not stay homogeneous once it cools.
The following lab period: Filter the crystals, wash them free of mother liquor with 10-15 mL of a cold 2:1 mixture of water and 95% ethanol, and air dry the solid for 15 min. Weigh your crude yield, break up any clumps of solid and recrystallize from hot 95% ethanol (8 mL per gram). You should not have to filter the hot solution. After cooling, the recrystallized benzoin should be filtered, washed with a minimum of a cold 2:1 mixture of water and 95% ethanol, and air dried for 15 min or left until the next lab period. Obtain the mp of the recrystallized benzoin (lit mp listed as 133 and 137 oC for d,l-benzoin, most students will see a mp of 133 oC) .
When you are satisfied that you have the product you want, you may dispose of the first filtrate by neutralizing with dilute HCl, then flushing the aqueous layer down the drain with plenty of water. The second filtrate (from recrystallization) can be flushed down the drain with water.
Step 2: Oxidation of Benzoin to Benzil
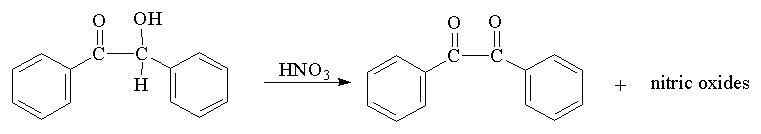
CAUTION: Concentrated Nitric Acid is extremely caustic and will burn exposed skin.
Work in a hood! Into a 125 mL Erlenmeyer flask, place 2.0 g* of benzoin (weighed to the nearest tenth of a g) and carefully add 7 mL of concentrated nitric acid. Heat the mixture on a steam bath with occasional slow swirling for 30 minutes or until the brown-red nitric oxide gases are no longer evolved. The fumes are toxic and noxious so be certain that the fume hood safety shield is pulled down.
Carefully cool the flask and contents using tap water (keep the flask covered with a plastic seal or a cork), then pour into 35 mL of cool water and swirl to coagulate the precipitated product. Collect the yellow solid using suction filtration and wash twice with 5 mL of cool water to remove some of the nitric acid present. Press the crystals to remove more water by placing another piece of filter paper over the crystals and pushing with a beaker or cork; the suction flask MUST be supported and sitting flat on the desktop. This crude product can be recrystallized from 95% ethanol while it is still slightly wet (4 mL/g). Dissolve it in hot ethanol, add water dropwise to reach the cloud point, and allow it to slowly crystallize. Filter, dry, record the yield, and take the mp.
* Do not use all of the benzoin you have synthesized. If you do not have more than 2.0 g, save 100 mg of the benzoin for a mp, IR, and to hand in and use the rest for this next step, altering the procedure to scale.
If your instructor requests, run a tlc of the recrystallized product with known samples of benzoin and benzil for comparison of Rf values.
When you are satisfied that you have the product you want, you may dispose of the filtrate by first neutralizing with sodium carbonate, diluting with water, and flushing down the drain. Ethanol from the recrystallization goes to the non-halogenated waste container.
Step 3: Preparation of Tetraphenylcyclopentadienone
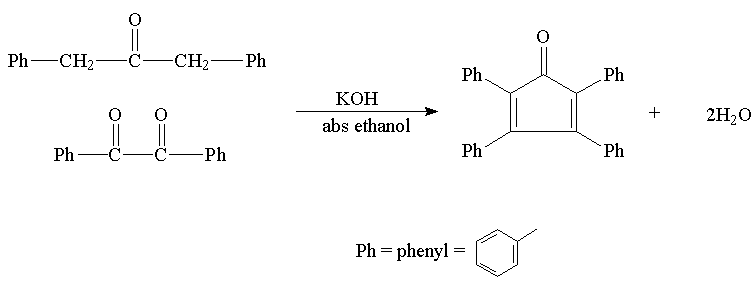
Cyclopentadienone is a relatively unstable compound which will dimerize even at low temperature. However, the corresponding tetraphenyl compound is quite stable.
Procedure: Into a 50 mL rbf, place 0.7 g* of benzil, 0.7 g of dibenzyl ketone, and 5 mL of absolute ethanol. Attach a reflux condenser and heat the mixture on a steam bath, water bath or sand bath until the solids dissolve. It is critical to prevent water (moisture) from coming into contact with the reactants. Raise the temperature to provide a slow reflux and add a solution of 0.1 g of potassium hydroxide (CAUSTIC) in 1 mL of absolute ethanol (this solution may be already prepared) dropwise through the top of the condenser. The reaction is very fast and a purple color will appear.
After addition of the base, allow the mixture to reflux for 15 minutes while periodically shaking the flask. Cool the reaction flask to room temperature, then in an ice bath. Filter using a Buchner funnel, wash twice with 5-mL portions of cold 95% ethanol, and air dry for an hour, if possible. When the crystals are dry (which may take until the next lab period), weigh, record the yield and the percentage yield . You may recrystallize a portion of the purple product using a 1:1 mixture of 95% ethanol and toluene (12 mL/0.5 g). Record the mp; literature mp 219-220 oC).
When you are satisfied that you have the product you want, the filtrate can be neutralized with dilute aqueous HCl and flushed down the drain. The recrystallizing solvent should be placed in the non-halogenated waste container.
*Do not use all of the benzil you have synthesized. If you do not have more than 0.7 g, save 100 mg of the benzil for a mp, IR, and to hand in and use the rest for this next step, altering the procedure to scale or obtaining additional benzil from your instructor.
Alternative Step 3: Reduction of Benzil with Sodium Borohydride
There are a wide variety of hydride reducing agents that convert carbonyl compounds into alcohols. One of the least reactive of these agents is sodium borohydride. Although it reduces aldehydes and ketones, it is fairly stable in aqueous and alcoholic solutions. The more reactive hydride reducing agents can reduce other functional groups such as carboxylic acids, esters, epoxides, and nitriles. Such hydrides react violently with water releasing hydrogen gas and must be handled very carefully. You will reduce the diketone, benzil, using sodium borohydride. Three stereoisomers of hydrobenzoin can be formed in this reaction, RR, SS, and RS which is the meso isomer. It is the meso isomer that predominates. The stoichiometry of the typical borohydride reaction is:
\[\ce{4 R_2C=O + NaBH_4} \rightarrow \ce{(R_2CHO)_4B}^-\ce{Na+}\]
hydrolysis of the borate ester
\[\ce{(R_2CHO)_4B}^-\ce{Na+} + \ce{H_2O} \rightarrow \ce{4 R_2CHOH}\]
Your starting compound is a diketone so you need one mmol of borohydride for every two mmol of ketone .
Procedure: Using a 25 mL or a 50 mL Erlenmeyer flask, dissolve 0.50 g of benzil (weigh to the nearest hundredth of a gram) in 5 mL of warm 95% ethanol. Cool the solution in a water bath which will produce a fine suspension of benzil particles. Add 0.10 g of sodium borohydride (weight to the nearest hundredth of a g) which will cause the solution to warm and dissolve the suspended benzil. As the reduction reaction proceeds in the next few minutes, the yellow color of benzil will disappear. After a total of 10 minutes, add 5 mL of water, heat to boiling on a steam bath, filter or decant if the solution is not clear. When the solution cools, dilute to the saturation point with as much as 10 mL of water and set the solution aside for crystallization to occur. In your discussion, mention that three stereoisomers are possible and suggest why the meso isomer (lit mp 136-7 oC) predominates.
When you are satisfied that you have the product you want, you may dispose of the filtrate by diluting with water, neutralizing the excess, unreacted borohydride with acetic acid, and flushing down the drain.
Step 4: Dimethyltetraphenylphthalate
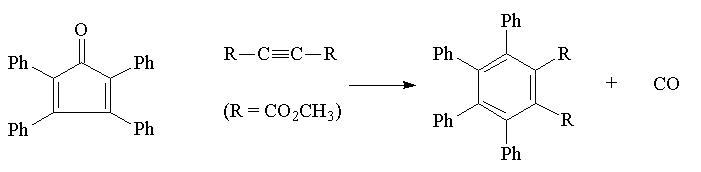
Into a 10 mL beaker, place 39 mg of tetraphenylcyclopentadienone (0.10 mmol), 3 drops of dimethyl acetylenedicarboxylate (an excess) and 0.3 mL of triethylene glycol. Mix the ingrediants by swirling, cover the beaker with a thin watch glass or appropriate microwave safe film (thick watch glasses often break in the microwave) and place it in the microwave oven. Set the oven at power level=6 for 5 minutes. After irradiating for 5 min, the beaker will be hot. Let it cool for a minute. The reaction mixture should be a golden color which cools to colorless crystals. It may take a few hours to see crystals. If necessary, leave the material to crystallize until the next class. Collect the crystals in a micro Hirsch funnel and wash with a few drops of cold 95% ethanol. Recrystallize using 95% ethanol. Record the product mp; literature mp 255-257oC.
Contributors and Attributions
James Chickos, David Garin, and Valerian D'Souza. University of Missouri–St. Louis; Chemistry)