
9.6: Experimental Procedure and Analysis
- Page ID
- 212008
VI. Experimental Procedure: Sample Preparation
General
The NMR sample tubes should be clean and free of dust and particulate matter. Solid samples should be dissolved in a deuterated solvent and liquid samples can be run neat or diluted in a deuterated solvent as described further below. A good 1 H NMR sample contains about 10 mg of compound. The solution should contain no solids or paramagnetic impurities. Your deuterated NMR solvent should be free of water, and your NMR spectrum should contain no solvent peaks.
An NMR sample tube is typically 175 mm in length with a 5 mm O.D. Minimum filling level is a distance of approximately 2 cm up from the bottom of the tube, which is equal to a volume of about 0.3 mL. However, the optimum filling level is a distance of approximately 4-6 cm up from the bottom of the tube (0.6-0.9 mL). You must determine the minimum height of a sample by checking the depth gauge in the NMR room. Over filling can distort the homogeneity and lead to lower resolution. When the tube is inserted into the probe, the position of the transmitter coil is about 1 cm from the bottom of the tube.
As mentioned above, the solvent must dissolve the sample material. It is generally desirable to use as concentrated a solution as possible -- i.e. about 10%. In difficult cases where the sample is not very soluble, it may be necessary to find an alternative solvent. Hydrogen-containing solvents should be avoided whenever possible. The selected solvent should not produce strong signals of its own in the spectral region of interest. Some solvents commonly employed for proton NMR spectroscopy include:
deuterochloroform -- CDCl3
deuterated water--D2O
hexadeuteroacetone (acetone-d6)--(CD3)2C=O
hexadeuterodimethylsulfoxide (DMSO-d6)--(CD3)2S=O
deuterodacetonitrile--CD3CN
deuterotetrahydrofuran--C4D8O
Chloroform is by far the most popular and will be used almost exclusively in 5.310. The TA will prepare the bottle of CDCl3 that you will use for your samples. The CDCl3 will be spectral grade already treated with TMS. Just keep in mind that you don’t want water getting into your deuterochloroform, so keep the bottle open to the atmosphere as little as possible. As long as the bottle remains open, water from the air will dissolve in your NMR solvent.
For preparing your NMR tube with an unknown liquid it is recommended that you take 50 uL of sample and dilute with 900 uL of deuterated chloroform in a small scintillation vial. Mix well and using a 9-inch glass pastuer pipette place the pipette into the NMR tube as far in as it will go then gently release the sample up to the tip of the pipette. The NMR tube should now be prepared perfectly with the correct volume of sample. The TA’s will demonstrate the technique for you during the pre-lab lecture. When your sample has finished running the NMR tube should be opened up and turned over into a glass beaker located under the hood, provided and labeled by the TA’s.
It is most important that the sample tube to be absolutely clean within and without; its surfaces must be completely dust-free. Particles just visible with the unaided eye may degrade the resolution if they are within the irradiation region.
Reference Material
It is usual to provide a reference line in each spectrum from which the chemical shifts of other signals can be measured. The normal method is to add a reference material (known as an internal standard) directly to the sample. The reference material should not interact with the test sample. If a symmetrical and non-polar type of molecule is used as the reference material, these effects will be very small. For this purpose, tetramethylsilane (TMS) is most suitable. An additional advantage of using TMS is that the spectral line it produces is at a higher field position than almost all other signals, so that the chemical shifts of nearly all lines measured with respect to TMS are of the same low field direction. As an alternative method, the reference material may be contained in a sealed capillary tube, which is inserted into the sample tube. There can be no intermolecular effects by this method. TMS will already have been added to the deuterochloroform, which we will be using.
Day 1 -Assembling a Ground-Glass Reflux Apparatus for Synthesis of an Ester, Extraction of Acidic Impurity from Product, Drying the Product.
The apparatus for synthesis of the unknown ester requires assembling a ground-glass reflux apparatus similar to that shown in Figure 5-1. Obtain from the stockroom a kit containing a 250mL ground-glass round bottom flask, 14/20 condenser, and an electric heating mantle which fits the 250mL round bottom flask. Secure an unknown organic alcohol and acid from your instructor and record the unknown number into your laboratory notebook.
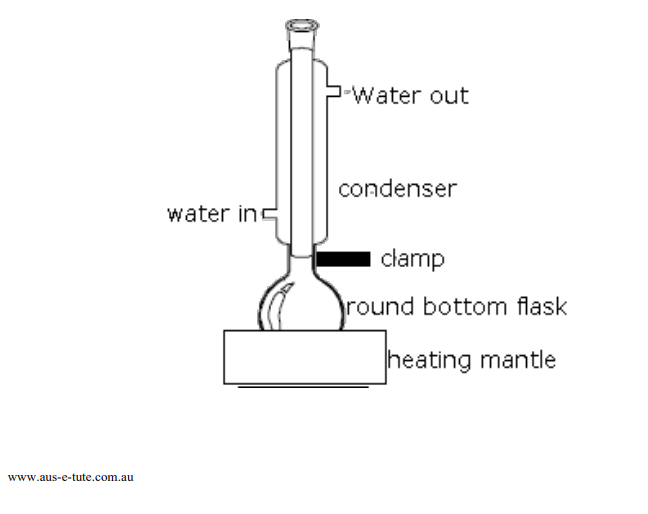
Figure 5-1 Reflux Apparatus Fischer Ester Synthesis
© AUS-e-TUTE. All rights reserved. This content is excluded from our Creative Commons license. For more information, see http://ocw.mit.edu/help/faq-fair-use/
When pouring the reactants into the 250mL round bottom flask do not allow the reactants to make any contact with the ground glass opening of the flask (See Figure 5-2). Rather, use a funnel this will keep the reactants from coming into contact with the ground glass opening of the flask. Both the sulfuric acid and the carboxylic acid are corrosive and should be handled carefully.

Figure 5-2 Use a Funnel for Pouring Reactants into the Reflux Flask
To a 250mL round bottom flask set up in a laboratory hood, add 40 mL of the unknown carboxylic acid followed by 30mL of the unknown alcohol. Gently, swirl the flask to mix the alcohol and carboxylic acid then, carefully add 3 mL of concentrated sulfuric acid H2SO4. Obtain the sulfuric acid from the small dropper bottles provided. Swirl the flask during the addition of the sulfuric acid the reaction is exothermic and heat will be released. Use caution in handling the acid it is highly corrosive and the liquid and vapors are harmful. Add a small magnetic stir bar to the flask, then, attach the reflux condenser with lightly greased joints as shown in Figure 5-3. Attach a drying tube to the top of the condenser.

Figure 5-3 Reflux Apparatus & Heating Mantle Fischer Esterification
Using a heating mantel connected to a rheostat apply heat (approximately 75 electrical units) bringing the solution to a boil, and reflux the solution for 1 hour. Objective is to achieve a slow rate of reflux with liquid boiling and condensed vapor dripping back into the flask. While the reaction is refluxing set up a 250 mL separatory funnel with a ring clamp support as shown in Figure 5-4.

Figure 5-4 Separatory Funnel with Ring Stand Support
Once the reflux has completed, cool reaction mixture to room temperature. Transfer the reaction solution to a 250 mL separatory funnel and add 60 mL of cold distilled water. Use an additional 20 mL of water to rinse out the reaction flask and add that to the separatory funnel as well. Swirl the solution, allowing the layers to separate and remove the aqueous layer. Pour the Organic layer into a 500- mL clean dry beaker. Extract the Organic layer with two 40 mL aliquots of 5% sodium bicarbonate solution to remove any traces of the carboxylic acid. CO2 gas is released during the extraction and this can cause a high pressure inside the separatory funnel. Because of this the first addition of 40 mL 5% sodium bicarbonate should be carried out in the 500-mL beaker slowly adding the sodium bicarbonate solution while stirring with a glass stirring rod. Once the fizzing subsides transfer the contents of the beaker back to the separatory funnel and continue with the remaining extractions. DO NOT INSERT THE GLASS STOPPER into the separatory funnel until the bubbling subsides. Test the second extracted aqueous layer with litmus paper and make sure that it turns litmus paper blue and is basic. If not, repeat the extraction with two more 40 mL portions of 5% sodium bicarbonate solution until the aqueous extract tests basic. Once the aqueous extraction layers have been removed extract the organic layer with two 10mL portions of saturated aqueous sodium chloride solution. The sodium chloride will help to pull any trace amounts of water out of the organic layer. Pour off the top organic layer into a 125 mL Erlenmeyer flask and dry with an anhydrous chemical drying agent such as MgSO4 or Na2SO4. Add approximately 4 grams of the drying agent swirling the flask gently, allowing the drying agent to clump up indicating that it is pulling water out of the sample. Add an additional 1-2 grams of the drying agent if needed until it stops clumping (caking) up in the flask. Typical set up is shown in Figure 5-5. After drying, the liquid should be very clear. Decant the organic layer into a clean scintillation vial and store it in your desk drawer until the next lab. Take great care to make sure that the drying agent stays in the original Erlenmeyer flask.

Figure 5-5 Drying Sample with an Anhydrous Drying Agent
Day 2 - Atmospheric Distillation
Set up a simple distilling apparatus as shown in Figures 5-6.

Figure 5-6 Atmospheric Distillation Apparatus with Vigruex Column
Now carefully, loosen the condenser clamp and disconnect the condenser and the distilling head from the 250mL distilling flask. Insert a funnel containing a small loose cotton plug into the 250mL distilling flask. Using the funnel, carefully pour the saved ester product into the clean dry 250 mL distilling flask. Add a magnetic stir bar to the distilling flask and re-attach to the distilling apparatus. Be sure that the adapter is open to the atmosphere. Have the system checked by your instructor prior to starting the distillation. Collect the distillate in labeled test tubes that are cooled in ice (See Figure 5-7). Fill a beaker with ice chips and use it to hold the test tubes during collection of the distillate. Make sure the test tube is positioned to capture any drops, which drip from the condenser.

Figure 5-7 Test Tube Receiver
Re-check that the cooling water has been turned on. When all has been checked, then carefully begin heating the distilling flask. For heating your apparatus start off at 40 electrical units on the rheostat control and go up 5 units every 5 minutes until you start collecting product. Try to aim for a distillation rate of approximately 1 drop of distillate every 2-3 seconds. Initially it may appear that nothing is happening, check to make sure the heating element is working and getting warm. Gradually as the heat is increased, condensation should begin to appear on the interior of the vigreux column and distilling head. Keep track of the thermometer readings as the vapor begins to condense off the tip of the thermometer bulb. At that point, the liquid should begin to collect inside the condenser and the first drop of distillate should reach your receiver test tube. At this point, carefully adjust the rate of heating on the rheostat such that you are collecting about 1 drop of distillate every 2-3 seconds. Continue at this heating rate and regularly record the temperature on the thermometer in the laboratory notebook. After collecting about 10-15 drops of distillate into the first test tube, change the collection flask by inserting and collecting into a second test tube. Collect the distillate in this second test tube as long as the temperature remains constant within a few degrees. If the temperature changes or if the test tube is more than half filled with product, change to another third clean test tube. Keep records of the temperature range that each fraction was collected at in the laboratory notebook. Make sure each of the tubes is clearly labeled in the order that product was collected. Continue the distillation until about 2-3 mL remains in the distillation flask. The clear colorless product should have an intense fruity odor. Under no circumstances should you distill your product to dryness. Once finished, shut off and unplug the rheostat from the wall, turn off the condenser water and allow the entire apparatus to cool to room temperature. Using one of the flat pan balances in the lab tear 150 mL beaker on the balance then place your first test tube receiver into the beaker recording the mass of the test tube and contents. Then, it might be good to also make a note on the odor of your product. Repeat this procedure for each receiver test tube that was used in collecting your product, recording the mass and odor of each fraction. Once the distillation apparatus is cool, disassemble the entire apparatus and wash and clean all of the parts with soap, hot water, and acetone air drying all components. Similarly, clean the separatory funnel and the reflux apparatus. Make sure all the glassware is absolutely clean and dry with no trace of any ester smell in the kit. Take the kit to the TA and have the TA initial the kit is being returned clean. Once you obtain the signature of your TA return the kit to the stockroom. Do NOT store the equipment in your desk drawers.
Day 3 - Characterize the Product by Infrared Spectroscopy (IR), Boiling Point (BP), Density, and Refractive Index (20nD) and NMR.
Day 3 will begin with the TA demonstrating the sample preparation and operation of the IR spectrometer, refractometer, and NMR spectrometer. Students will then prepare an NMR sample tube and run their spectrum on the 60MHz NMR spectrometer. Following this student’s will proceed to determine the BP, Density, Refractive Index, and run an IR spectrum on the Unknown Ester.
The thermometer calibration part of the experiment is performed to check the accuracy of the digital thermometer or regular thermometer that will be used in this experiment. The boiling point and freezing point of water can be used to calibrate the thermometer for this experiment. Take two readings on the thermometer one for the freezing point of water and one for the boiling point and compare those readings with the true values. For the freezing point fill a 150 mL beaker with ice and add distilled water. Let it sit for 10-15 minutes until it’s cold then take the temperature of the ice water mixture. Record the measured value and note the theoretical value, which should be 0°C. While waiting for the water to get cold fill a 150 mL beaker about ¾ full with water and heat it up on a hot plate to boiling (cover the beaker with aluminum foil to speed up the process). This may take 20 minutes. While the water is heating, take a reading for the barometric pressure in the lab from the barometer in inches of mercury. Do a quick calculation to find the correct theoretical boiling point of water at that pressure. Temperature corrections to the boiling point of water can be calculated using the following formula:
Theoretical B.P. correction = (atmospheric pressure – 760 mm Hg) x (0.037°C / mm)
Theoretical B.P. = 100 °C ± correction above
Check the temperature of the boiling water as it is heating up. Be sure not to touch your thermometer to the sides or bottom of the beaker. Record the experimental boiling point of the water. Using excel, construct a thermometer-calibration curve by plotting observed temperature values versus true theoretical values.
Boiling point can be measured very accurately using an electronic digital thermometer and the apparatus pictured in Figure 5-8a,b. This device can be used to measure the boiling point of our unknown organic liquid. Check with the TAs to see if this reaction should be running under the hoods. Pour about 0.3 mL of liquid into a 12 x 75 mm test tube. Add a boiling chip to the test tube. Note in particular the position of the thermometer probe, which should be placed about 5 mm above the liquid and is positioned so that it does not touch the test tube at any point. The test tube sits in an 80 x 40 pyrex heating dish filled with sand, which is heated by an electric hotplate. Heat the sand until the liquid boils gently in the test tube so that the vapor is condensing about 3 cm from the top of the test tube but gently enough so that the liquid does not boil out of the apparatus. After a few minutes, you should begin to see condensation on the thermometer tip and droplets falling from the thermometer element. After a few more minutes the temperature should become reasonably steady. This is the boiling point of your volatile unknown organic liquid record it into your laboratory notebook. Avoid unnecessarily breathing and inhaling the vapors from your system the unknown liquids should be assumed to be flammable and toxic.

FIGURE 5-8a Photo of Boiling Point Apparatus
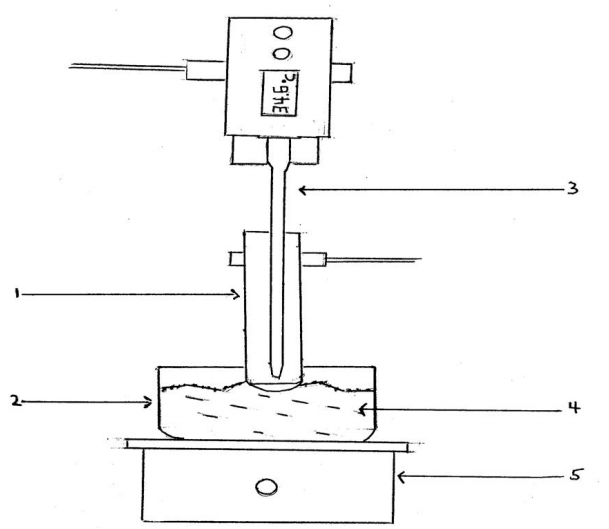
Figure 5-8b Schematic Drawing of Boiling Point Apparatus
1. 13x100mm test tube
2. 80x40 pyrex heating dish
3. digital thermometer
4. white sand
5. electric hot plate
To determine the density of the unknown pour 10 mL of the unknown liquid into a clean dry 10 mL graduated burette. The easy way to do this is to use a flexible plastic Pasteur pipette do not use a glass pipette as it could break and clog the burette tip. It should only take 2-3 fills of the plastic pipette to fill the burette with unknown. The hole on a 10mL burette is too small to try pouring the liquid in with a beaker. Use the plastic disposable pipette. Dispose of the plastic pipette in the solid waste container not the glass box. Determine volume readings of the liquid to ±0.01 mL then take mass readings of the liquid on a Sartorius balance to ±0.001 g. Place a clean glass weighing bottle on a top-loading Sartorius balance and cover with the aluminum balance cover so that the head of the bottle just sticks out the covers opening. Tare the balance to zero. Using a burette clamp, carefully add approximately 1.50 mL of unknown liquid to the weighing bottle and immediately record the mass in your lab notebook. Cover the top of your weighing bottle & the top of the burette with a plastic weighing boat while you take the volume reading from the burette. The plastic weigh boat fits perfectly over the bottle and burette and will prevent any of the volatile liquid from evaporating. Record the volume reading into your lab notebook. Remove the plastic weighing boat cover the weight should be the same to within a few thousandth of a decimal place. Now add another 1 to 2 mL of liquid to the weighing bottle and follow the same procedure recording the total volume and new mass. Continue this until six or seven data points are collected. Make sure to take the last mass reading after adding around 9.50 mL or so of the unknown. Enter data into an excel spreadsheet and create a graph of data plotting mass on the y-axis versus volume on the x-axis. Find the best least squares line through the data points with an equation for the line. Make sure when adding a trend line that you set the intercept at zero, and display the equation in chart. The equation gives you the slope of the line, which should be the density of the unknown volatile liquid. For the molar mass and density do a detailed error analysis with 95% confidence limits.
Measure the refractive index of the sample following the procedure demonstrated by your TA and outlined in Appendix IV. Remember, refractive index varies inversely with temperature (T). Make sure to record the temperature on the thermometer in the refractometer. You can then make an adjustment to the refractive index when comparing with a literature value that may have been obtained at a different temperature. Use the following formula for calculating the correction: Δn = 4.5E-4 (T1-T2) where T1= temperature of the refractometer and T2= temperature you are comparing to. Then, add the correction to the measured refractive index obtained from the instrument. The Abbe Refractometer is shown below in Figure 5-9.

Figure 5-9 Photo of Auto Abbe Refractometer
NMR Sample Tube Preparation: Obtain a clean dry 5mm NMR tube from the stockroom. Check the tube carefully for any cracks and/or chips. Obtain a bottle of deuterated chloroform (CDCl3) from the stock room. Dissolve 50 µL of the ester in 900 µL of CDCl3. If the solution is clear add the solution to the NMR tube using a 9” glass Pasteur pipette. Insert the pipette filled with your sample into the NMR tube as far as it will go, then release the sample until it reaches the opening (tip) of the pipette. At that mark withdraw the glass pipette and the NMR sample tube now contains the correct amount of liquid. If the solution is cloudy filter using a small piece of kim wipe inserted into a glass Pasteur pipette. Cap the NMR tube and label with your name. See Appendix III for the operation of the NMR 60.
Day 4 - Identification of the Unknown Ester followed by running a Mass Spectrum
From the observed boiling point, density, refractive index and NMR spectrum of the unknown ester, students will fill out the Preliminary Identification of Unknown Ester form and turn it in to your instructor at the start of the lab on Day 4. The first identification is worth 5 points. Once the identification form has been turned in to your instructor, run a mass spectrum on the previously identified ester. Then, fill out the final Identification of Unknown Ester form and turn it in to your instructor prior to the end of lab on Day 4. The second identification is worth an additional 5 points. If it is correct you are ready to begin writing your report. If it is not correct you may try to determine the source of your error and verify it with your TA. All of the laboratory work must be completed in the allotted four days for this experiment and incorrect attempts at identification in no way affects the written report due date. If you missed the identification you will be given the name of the correct ester for work up and analysis in your report.
Mass Spec Sample Preparation: Obtain a small scintillation vial from the stockroom. Dissolve three drops of the ester in 1 mL of Pentane. Proceed to the Mass Spectrometer and inject 0.01 µL of your sample into the GC-MS. Each run takes 10 minutes + 3 minutes for data analysis. Your TA will have a sign-up sheet for the GC-MS. Please follow the direction of the TA.
Mass Spec Sample Preparation: Obtain a small scintillation vial from the stockroom. Dissolve three drops of the ester in 1 mL of Pentane. Proceed to the Mass Spectrometer and inject 0.01 µL of your sample into the GC-MS. Each run takes 10 minutes + 3 minutes for data analysis. Your TA will have a sign-up sheet for the GC-MS. Please follow the direction of the TA.
Analysis and Discussion
- Indicate the structural formula of the ester and how it was obtained from the proton NMR. Describe how the structure is consistent in terms of the proton NMR spectrum. Compare the calculated chemical shifts with the actual values obtained from the NMR. If you missed the identification of the unknown ester, present reasoning and indicate what it should have been in terms of being consistent with the data collected.
- Summarize the data in table form that indicates the identity of the ester. Talk about the IR spectrum and what peaks were present/absent that indicated an ester. Which features of the IR spectrum can be assigned to the ester? Be sure to identify and label as many functional groups as you can.
- Compare the boiling point, density, refractive index and composition of the ester identified from your data with literature values. Explain any discrepancies.
- Comment on the quantities of alcohol/acid used in the experiment as well as the sulfuric acid in terms of Kinetics and/or Thermodynamic reasoning. Discuss the yield of the product in terms of both of the above. How would each of the following shift the equilibrium in the esterification reaction (a) removal of H2O as its formed (b) adding more H2SO4 catalyst.
- Write out the synthesis steps for the ester that you identified. Include all of the names, structures, catalysts, products and reaction conditions that were used in the synthesis. Identify the limiting reagent and show calculations including a table or results. Calculate the theoretical yield and percent yield of product formed.
- Comment on the odor of the ester (this should be carefully obtained and you should not inhale or breathe the ester directly but waft the air to obtain the scent. Research the ester and determine if the smell observed was accurate in addition describe clearly where this ester is found and how it is used. What are the association of acid/alcohol and the odor of your ester?
- Talk about sources of error in this experiment and how the experiment could be improved.
- Aside from any improvements in experimental techniques used in this experiment describe how the % yield of the product might be increased.
- Write out the full mechanism (with arrows) for the formation of the ester that was synthesized.
- Analyze the IR spectrum by reporting the observed bands in a table and assigning (labeling) those bands on the spectrum. Identify and label as many functional groups as you can.
- Analyze the NMR spectra by reporting tabulated results and assigning (labeling) those peaks to H atoms in the identified molecules. Identify, label and explain all of the peaks.
- Analyze the MS spectrum reporting on the major peaks and assigning (labeling) those peaks.
- Your well written discussion should address the following (within the text, NOT as a numerical series of answers to questions):
- Comparison of your distillation results (temperature ranges) fractions collected with your GC results. .
- Explanation of how the separation of components could be improved.
- Comparison of your results with literature values (boiling points/compositions/yields)
- Discussion what information gas chromatography provides.
- Discussion what information infrared spectroscopy provides.
- Discussion what information mass spectroscopy provides.
- Discussion what information nuclear magnetic resonance spectroscopy provides.
- Describe the odor of your carboxylic acid and alcohol prior to the reaction