
13: Synthesis of Vitamin B₁₂
- Page ID
- 21852

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)The total synthesis of Vitamin B12 was accomplished in 1973 by a grand collaboration between R. B. Woodward’s group at Harvard University (USA) and A. Eschenmoser’s group, Swiss Federal Institute of Technology (ETH), Zürich, Switzerland. It took about twelve years and more than two dozen senior scientists to complete this gigantic task. The achievement is variously eulogized by organic chemists – monumental achievement in the annals of organic synthetic chemistry; a breakthrough; a mile stone in organic synthesis; unrivaled even after 40 years; a task no less adventurous than concurring Mount Everest. At the time the adventure started it was the most formidable challenge in synthetic chemistry, which few would have dared. The announcement of its synthesis marked the coming of age of synthetic organic chemistry. Woodward and Eschenmoser worked in close collaboration and in competition during this historic pace. In the process, they achieved not only an astounding synthesis, but also opened several new fields for future investigations. Woodward Hoffmann Rule is the most famous of the offshoots. While studying this synthesis, a student should also ponder over the thoroughness in their planning of all aspects of the scheme and initiation of suitable basic studies well in advance to facilitate the main scheme at appropriate junctures. This long introduction is just in tune with the length of the scheme, time taken and magnificent achievements.
Retroanalysis of Vitamin \(B_{12}\) synthesis
Figure 13.1 shows the structure of Vit B12 and the main structural features / challenges of the complex molecule.
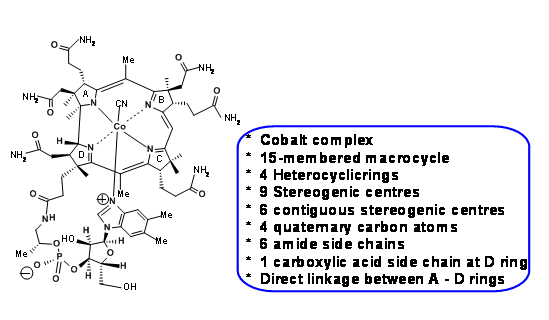
In his inimitable style, R. B. Woodward draws attention to these challenges in the beginning of his lectures and to the fact that it took close to 50 years to establish the structure of Vit B12 by chemical degradation and finally by X-Ray diffraction studies by Dorothy Hodgkin in 1956. One should note that most of the chemical degradation studies, remarkable as they might be, happened during the early years of the twentieth century, when modern organic chemistry was in its making. Spectroscopic data, if any, were conspicuous by their absence and most of the degradation and synthetic chemistry were too harsh by modern (meaning 1960s) standards for this delicate molecule. Hence, this was a daunting scenario. This meant that a whole lot of synthetic chemistry had to be reinvented to suit these challenges. Such aspects were thoroughly planned in and contingency plans were put in place well in advance. Discerning students could see a glimpse of this planning in this short presentation. The ideas on retroanalysis, as we understand today, were in the developing stages during the sixties. The retroanalyses we use here are later-day additions by other scientists, based on the facts (lectures, papers etc.,) published by the two groups. All such citations are included at the end of the write-up.
The synthetic target was identified as Cobyric Acid, because this compound was a natural product and had been converted to Vit B12 by Bernhauer K., et.al., (1960)(Figure 13.2). Hence total synthesis of Cobyric Acid would amount to a formal synthesis of Vit B12.
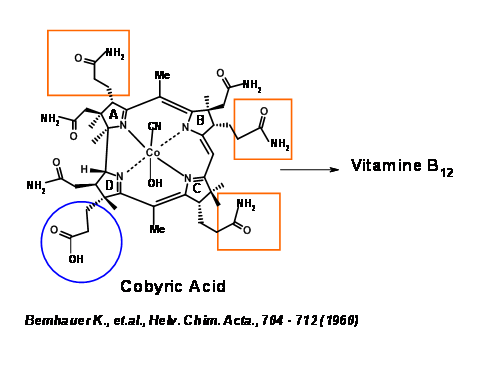
Cobyric acid had seven carboxylic acid side chains, out of which four were propionic acid moieties, one on each heterocyclic ring. The main challenge was to differentiate the propionic acid chain on the D ring from the other acetic acid and propionic acid moieties. It was therefore decided that this odd acid moiety would be masked as nitrile (Figure 13.3). That still leaves a daunting task of differentiation, which we could address later. It was first decided to view the molecule as made up of two halves – the Eastern Side and the Western Side. The first disconnection was at the A/B ring junction at the methylene bridge (1.13.3A). Cleavage of the second bridge at C/D ring junctions gave the Eastern Half as Thiodextrolin in charge of Eschenmoser’s group at Zurich and the Western Half as Cyanobromide in charge of Woodward’s group at Harvard (US).
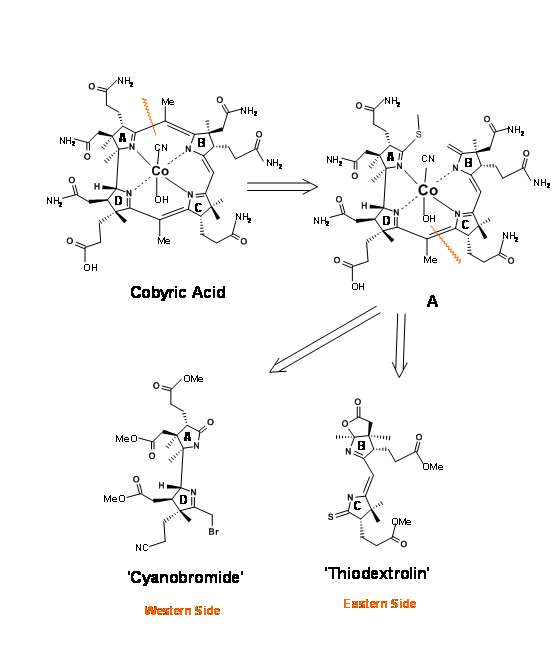
Retroanalysis of cyanobromide
This western half has a formidable array of six contiguous stereocentres on an eight-carbon frame. Note that the stereocentres were planned on the basis of known stereoselectivities and the six membered rings were built to provide the propionic acid chains (Figure 13.4). The nitrogen for the A ring came from an indole, whose benzene ring gave the side chains for the A ring. The D ring nitrogen came via a Beckmann rearrangement. Corrnorsterone (1.13.4A) was the key intermediate (corner stone) that held all the stereocentres and chains on the Western Half.
Synthesis of Western Half
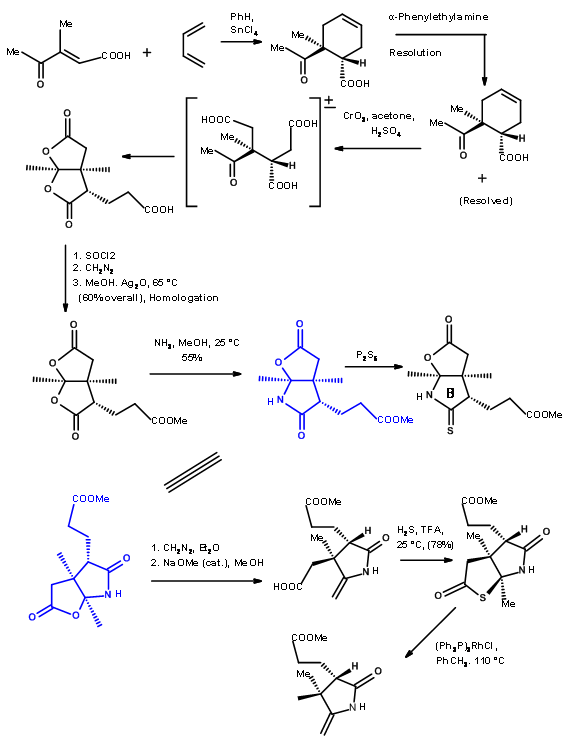
The required enantiopure 1,2,3-trimethylyclopentene unit came from camphorquinone as shown in Figure 13.5.

Through another convergent synthesis, a five membered ring was fused to indole at C2 – C3 bond and resolved as shown in Figure 13.6.
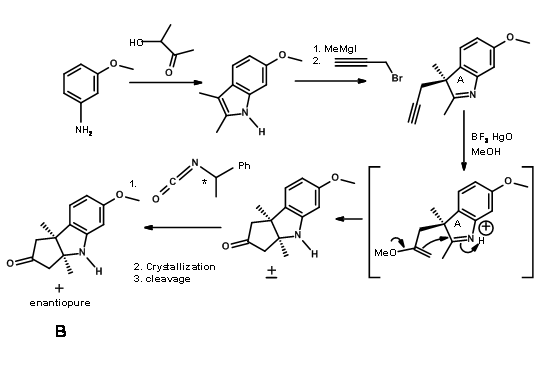
The (+)- enantiomer was actually needed for target synthesis. The useless (-)- enentiomer was used as a model compound (for this was "just about the only kind of model study which we regard as wholly reliable” – RBW).
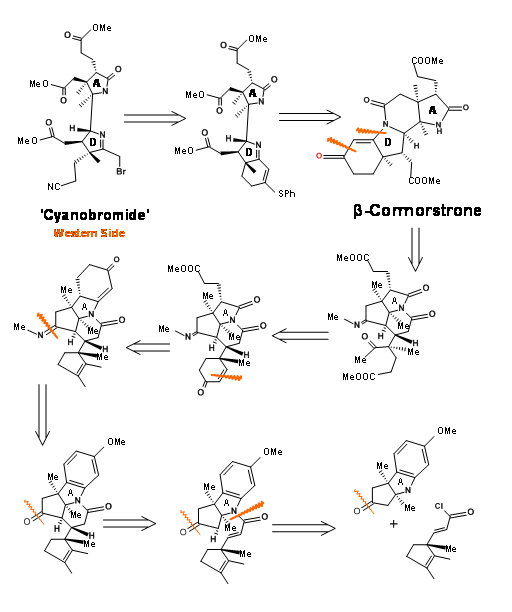
Fragments A and B were combined and then processed to Corrnorsterone as shown in Figure 13.7.
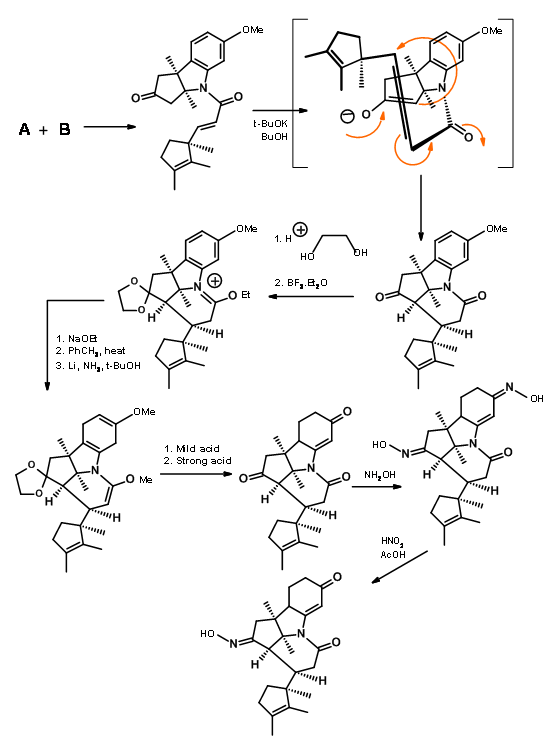
Note that the Beckmann ring expansion process set in motion a cascade of reaction, leading to a Claisen condensation and D ring formation, all in one step. The six membered imide carbonyl was also cleaved and placed an acetate chain on the D ring. In spite of this elaborate planning and execution, the process yielded a mixture of epimers at propionic acid side chain on the A ring, the required isomer being a minor component of the mixture. The major undesired product does not cleave at the amide bond due to unfavourable steric compression at the developing side chains (Figure 13.8).
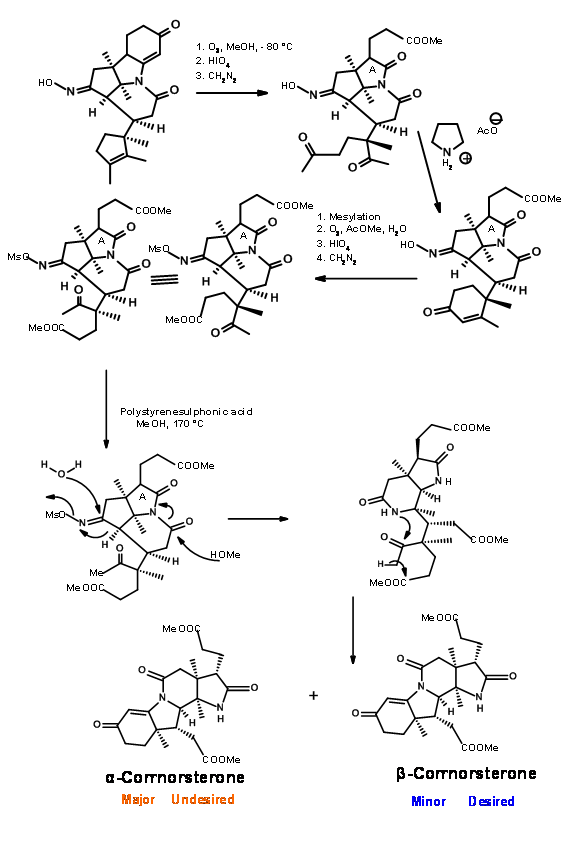
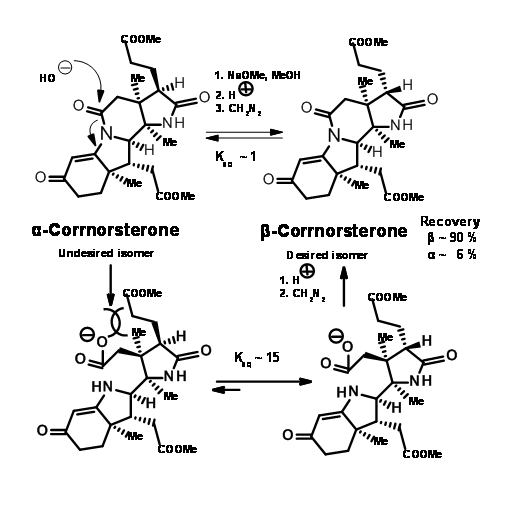
This unfavorable steric problem was however soon solved. On hydrolysis under strong base conditions, the amide ring opened and the propionic acid side chain isomerised to the less strained isomer. This could be then be acidified and esterified to β-corrnorsterone, with a recovery of 90% of the desired isomer (Figure 13.9).
This correct isomer was treated with a mixture of methanol and thiophenol under acid conditions (Figure 13.10). This set two processes in motion. The thiophenol attacked the ketone and activated this center, while the methanol oxygen attacked the amide bond leading to an ester and a thioenol ether.
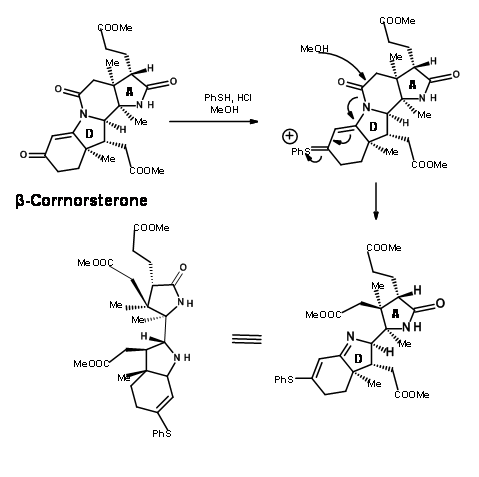
Ozonolysis of the thioenol ether at – 90°C cleaved the olefin unit to the aldehyde-thioester compound (Figure 13.11). An interesting new chemistry evolved here. While thioesters are less reactive to acid hydrolysis and showed comparable reactivity with oxygen nucleophiles, nitrogen nucleophiles were unique. The thioester reacted much faster then normal oxyesters to give amides. Thus, the thioester was exclusively cleaved to amide with ammonia, leaving three methyl esters untouched. The aldehyde moiety was then selectively converted to alcohol and then mesylated under mixed anhydride / pyridine conditions. This sequence also converted the amide moiety to nitrile. The mesylate was then converted to bromide to give the key intermediate cyanobromide.
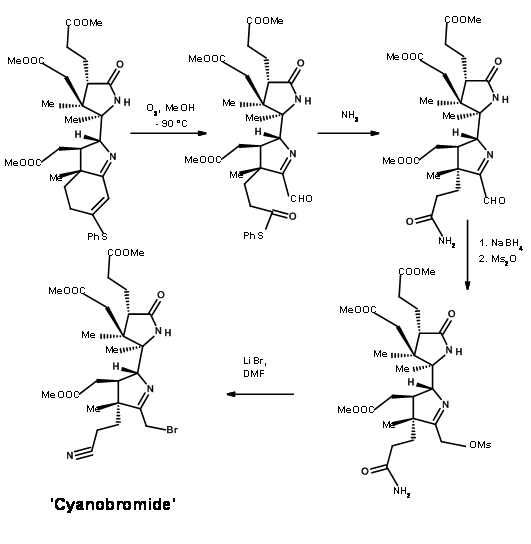
The Zurich group was simultaneously working on the synthesis of the Eastern Half named Thiodextrolin. The fragments for B and C rings were planned via., a single intermediate (1.13.12).
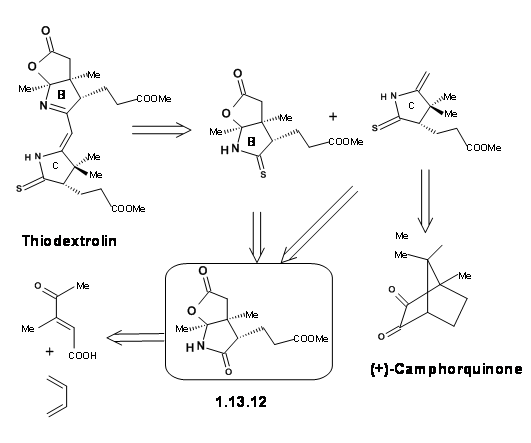
The synthesis started with a Diels-Alder reaction to secure two asymmetric centers properly and the racemate was resolved. The pure enantiomer was followed through the scheme to obtain the B ring segment. The same intermediate yielded the C ring fragment as well (Figure 13.13).
To connect two such fragments Eschenmoser had developed two sulphide contraction procedures. The mechanisms of the processes are shown below.
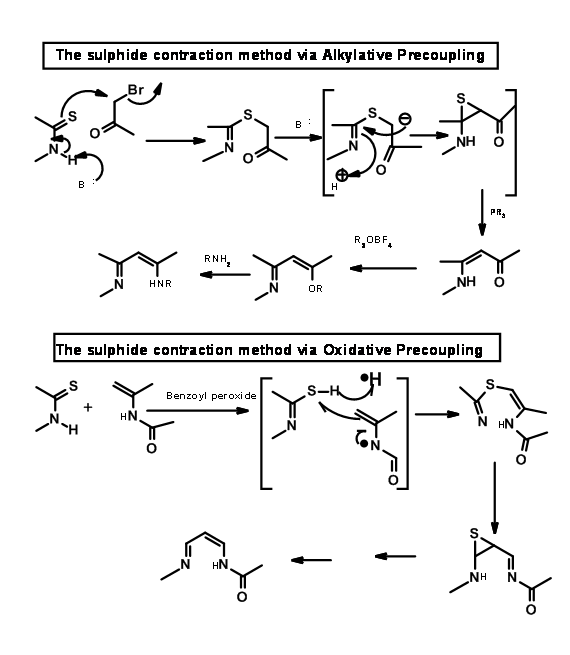
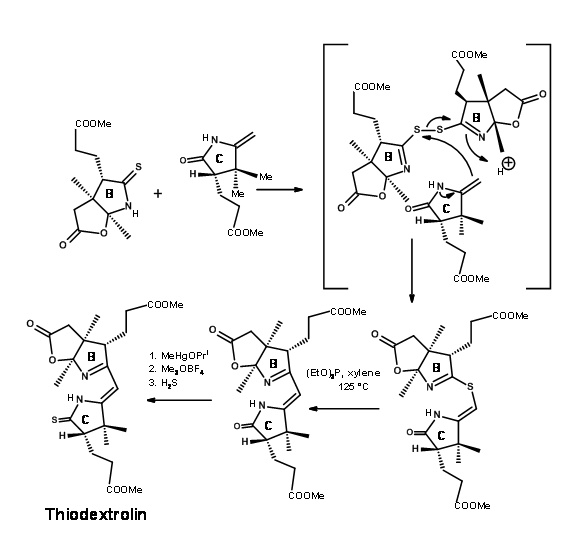
Using the oxidative coupling / sulfur extrusion procedure, they coupled the B and C rings as shown in Figure 13.15.
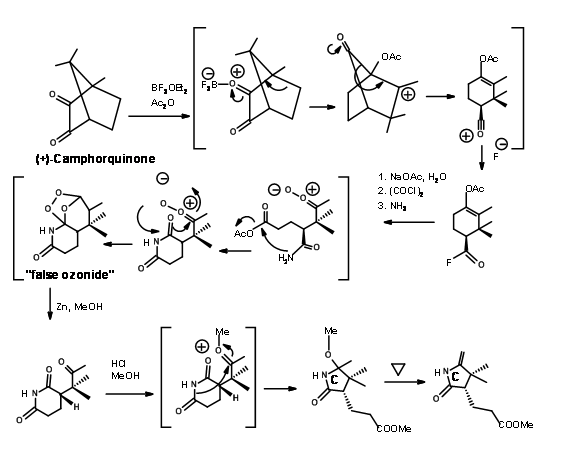
Woodward’s group also developed a new synthesis for the C ring starting from (+)-Camphorquinone. The scheme is shown in Figure 13.16.
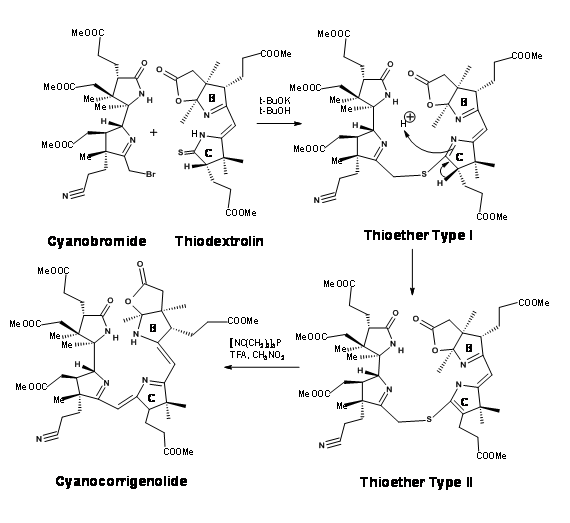
Though great care had been spared for the stereocentres at all points, the problem of stereoisomers could not be avoided. The crystalline thiodextrollin that was synthesized was actually a mixture of two stereoisomers at the propionic acid moiety of the B ring. Though they had purified the mixture at this point, it was of no value because this stereocentre was due for further disturbances at later stages. The mixture was taken forward for the first coupling at C / D bridge. After considerable effort that lasted over a year, they were first coupled at the Southern end using the alkylation / extrusion procedure (Figure 13.17). Note that the first product of alkylation was a thioether Type I, which readily isomerised to thioether Type II. The product was named Cyanocorrigenolide. This isomerisation disturbed the stereocentre at the C ring.
The next phase was the formation of A / B bridge. The C / D coupled compound was first treated with phosphorous pentasulphide followed by trimethyloxonium fluoroborate (Me3OBF4) (Figure 13.18). This procedure replaced the oxygens on the A and B rings by sulfur and finally to the S-methyl derivative. Dimethylamine in methnol cleaved the thiolactone ring selectively to a dimethylacetamide chain and a terminal olefin. The olefin was rather unstable. This was to be converted immediately to the cobalt complex. This procedure was not easy. Under several conditions, the cobalt metal ion catalyzed further reactions leading to extensive “destruction” of the compound. After several experiments it was observed that cobalt chloride or iodide in THF was unique for smooth cobaltation. The complexation process brought the A and B rings in close proximity. A base catalysed reaction then enabled formation of the bridge and removal of the sulfur moiety the best condition being DBN catalysed cyclization.
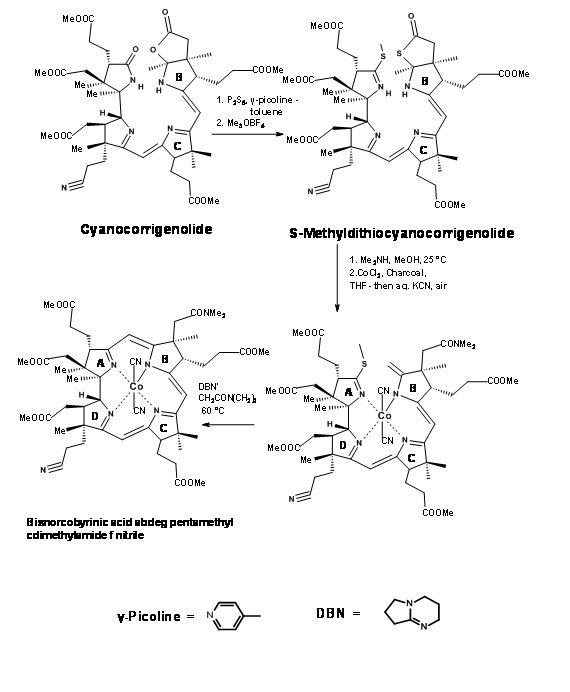
Note that the overall transformation interfered with the asymmetric center at C ring. Nonetheless, the A / B bridge was finally in place.
The Zurich group also came up with an alternate Zn-complex procedure along the same lines. All these manipulations were indeed harsh to the (three) epimerisable propionic acid chains on the A, B and C rings. The final product was purified by TLC (“plate chromatography”) and critically analyzed by HPLC (a new chromatographic tool at that time). The UV chromophores in all the products were of great help in this chromatography.
The synthesis has now two major milestones to pass. There are three active methine bridges in the molecule. The bridge at B / C rings had to be ‘protected’ from methylation. This was achieved by an oxidative lactone formation reaction at the B ring (I2, AcOH) (Figure 13.19). This new quaternary center and the existing quaternary carbon at C12 together exerted a steric congestion around C10. The chloromethyl ether entered the C5 and C15 centres exclusively. Ra-Ni hydrogenolysis cleaved the thioethers and the lactone ring in one step. This step was followed by esterification.
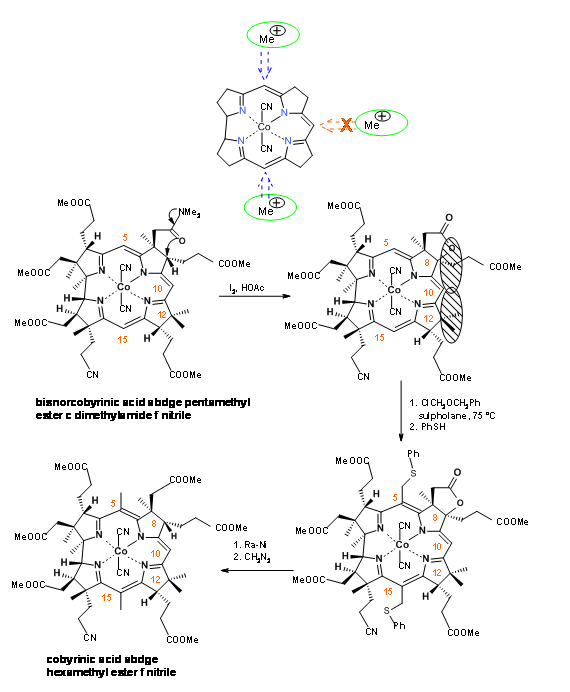
Conc. sulfuric acid converted the nitrile to amide (Figure 13.20).At this stage, they faced the difficult task of selective cleavage of amide, in the presence of six esters in the molecule. After extensive parallel experimentations, the Harvard group rediscovered an efficient selective cleavage of the amide moiety in N2O2 reagent.
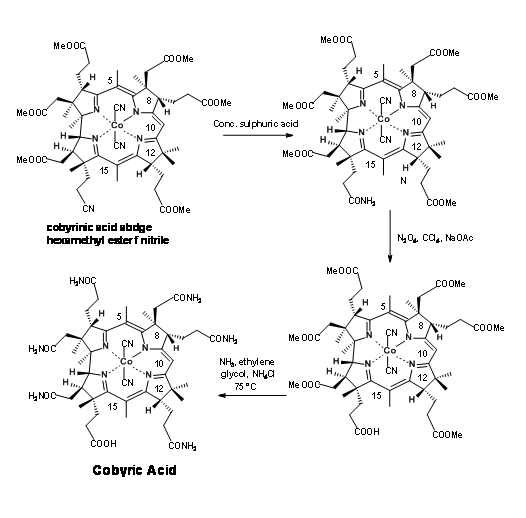
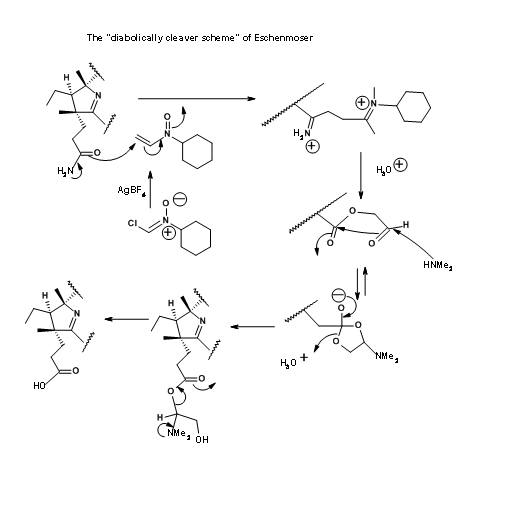
The Zurich group also came out with a “diabolically cleaver scheme” for selective hydrolysis of the amide group in the presence of ester groups. This scheme is shown in Figure 13.21. However the former was preferred due to its simplicity and better yields. Nonetheless, Eschenmoser’s solution stands testimony to human ingenuity.
The very last step of this long synthesis posed a major problem that needs special mention. Amidation of esters with ammonia was the only remaining step for the final assault on the synthesis of Cobyric acid. A closer look reveals that the task may not be that easy. The ester moieties (in particular the acetate units) were in crowed environments. Parallel to these developments, model studies on very similar molecules were in progress. Based on these studies, when the hexamethylester 1 acid was treated with ammonia in ethylene glycol at 75°C for 30 hours, the product obtained was not cobyric acid but a pseudocobyric acid, whose structure was established as dehydrocobyric acid. This product could be purified only by HPLC. This was one among the products obtained by earlier workers. But they had no idea about such a complication. This mystery took several critical studies to solve. Throughout all these studies, great care was exercised to see that the solvents were well deoxygenated before use. Oxygen was strictly avoided from the reaction atmosphere. The source for this oxidative cyclization was attributed to cobalt, which was suspected to oxidatively bond to the C9 position, facilitating the amide anion to cyclise. This unwanted reaction took a long time to solve. Several studies were aimed at ammonialysis under reducing conditions and also to open the lactam ring under reduction conditions.
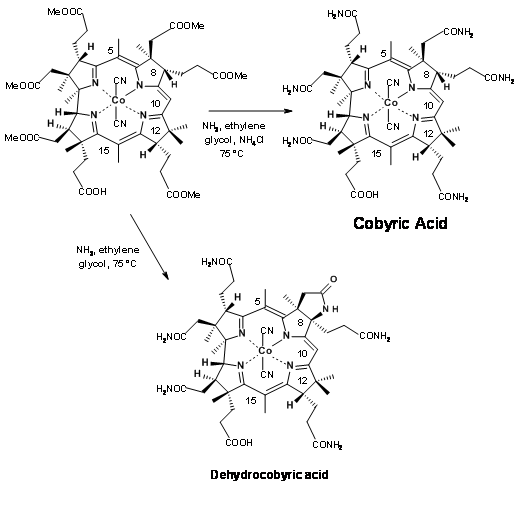
Finally a few milligrams of ammonium acetate were added to the reaction mixture to prevent the formation of amide anion, which was suspected to be the culprit for cyclization (Figure 13.22). This trick helped. The reaction was complete within 10 hours, with cobyric acid as the only product in good yields. This cobyric acid was identical in all respects, particularly in HPLC, with the natural product, thus ending this long journey for the formal synthesis of Vitamin B12.
This extraordinary adventure in organic synthesis is noted for several most significant achievements.
- Developments in Corrin chemistry
- Synthetic strategy
- Development of several new methodologies
- Woodward-Hoffmann Rules
Even after half a century this Vit B12 synthesis remains unrivaled and continues to inspire generations of chemists.
Further Reading
- Recent advances in the chemistry of natural products, Woodward, R.B. Pure & Appl. Chem., 17, 519 (1968).
- Eschenmoser, A. Pure Appl. Chem., 297–316 (1963).
- Eschenmoser, A. Angew. Chem. Int. Ed., 5 (1988)
- Recent advances in the chemistry of natural products, Woodward, R.B. Pure & Appl. Chem., 25, 283 (1971).
- The total synthesis of vitamin B12, R. B. Woodward, Pure & Appl. Chem., 33, 145 (1973).
- Natural Product Synthesis and Vitamin B12, Eschenmoser, A.; Wintner, C.E. Science, 196, 1410 (1977).
- Crowfoot-Hodgkin, D. et al. Nature, 178, 64 (1956).
- Friedrich, W., Eds., Walter de Gruyter: Berlin, 37, (1979).
- Targets, Strategies, Methods. Weinheim, Germany: VCH, (1996).
- The Art and Science of Total Synthesis at the Dawn of the Twenty-First Century, K. C. Nicolaou, Dionisios Vourloumis, Nicolas Winssinger, and Phil S. Baran, Angew. Chem. Int. Ed., 39, 44 (2000).
- Vitamin B12 : An Epic Adventure in Total Synthesis, by Neil Garg, January 29, 2002.
- The Asymmetric Total Synthesis of Vitamin B12, Nathan S. Werner, Denmark Group Meeting, September 28th, 2010.
- Nicolaou, K.C.; Sorensen, E. J. Vitamin B12. Classics in Total Synthesis, VCH: New York, 100, (2003).