
13.2: Isomerization reactions
- Page ID
- 978

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)Enols, enolates, and enamines are reactive intermediates in many isomerization reactions. Isomerizations can involve either the interconversion of constitutional isomers, in which bond connectivity is altered, or of stereoisomers, where the stereochemical configuration is changed. Enzymes which interconvert constitutional isomers are called isomerases, while those which catalyze the interconversion of enantiomers and epimers are called racemases and epimerases, respectively.
13.2A: Carbonyl isomerization
Two important chemical steps in the glycolytic pathway, catalyzed by the enzymes phosphoglucose isomerase and triose phosphate isomerase, involve successive keto-enol tautomerization steps. In both reactions, the location of a carbonyl group on a sugar molecule is shifted back and forth by a single carbon, as ketones are converted to aldehydes and back again - this is a conversion between two constitutional isomers.
Let's look first at the triosephosphate isomerase reaction, in the ketone to aldehyde direction. The ketone species, dihydroxyacetone phosphate (DHAP) is first converted to its enol tautomer with the assistance of an enzymatic acid/base pair (actually, this particular intermediate is known as an 'ene-diol' rather than an enol, because there are hydroxyl groups on both sides of the carbon-carbon double bond). The initial proton donor is positioned in the active site near the carbonyl carbon, and significantly lowers the pKa of the alpha-proton.
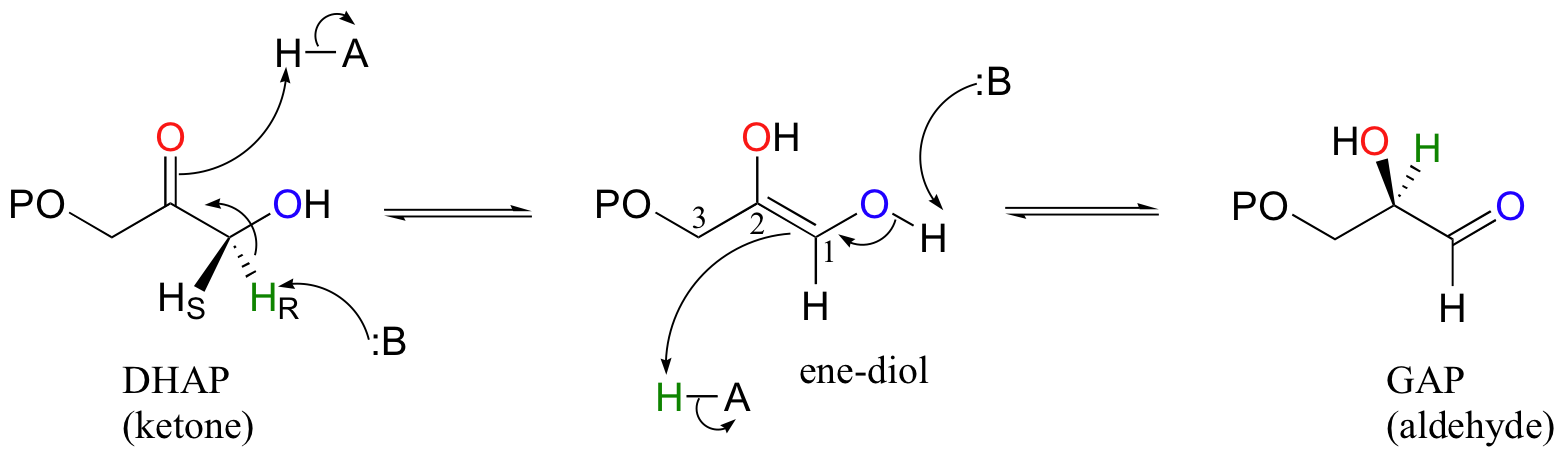
The second step, leading to glyceraldehyde phosphate (GAP), is simply another tautomerization, this time in the reverse direction. However, because there happens to be a hydroxyl group on C1, the carbonyl can form here as well as at C2. Notice that DHAP is achiral while GAP is chiral, and that a new chiral center is introduced at C2. The catalytic base abstracts the pro-R proton from behind the plane of the page, then gives the same proton back to C2, again from behind the plane of the page.
In the phosphoglucose isomerase reaction, glucose-6-phosphate (an aldehyde sugar) and fructose-6-phosphate (a ketone sugar) are interconverted in a very similar fashion.

The enzyme ribose-5-phosphate isomerase (EC 5.3.1.6), which is active in both the Calvin cycle and the pentose phosphate pathway, catalyzes an analogous aldehyde-to-ketone isomerization between two five-carbon sugars.
13.2B: Stereoisomerization at chiral carbons
The enolate form of a carbonyl is a common intermediate in a type of stereoisomerization reaction known as an epimerization. Recall from section 3.7A that the term 'epimer' refers to a pair of diastereomers that differ at a single stereocenter. The five-carbon sugar phosphates ribulose-5-phosphate and xylulose-5-phosphate, for example, are epimers: they are identical except for their stereochemistry at C3. A reaction in the Calvin cycle, which plants use to incorporate, or 'fix', the carbon atom from carbon dioxide into sugars, involves the interconversion of these two sugar phosphate epimers - in other words, inversion of configuration at C3.
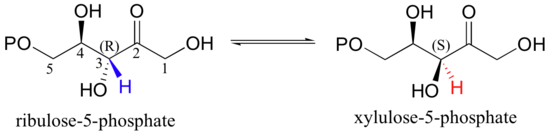
The reaction is simply a deprotonation at C3 to form an enolate, followed by reprotonation to return to the ketone.

The key to epimerization is that deprotonation and reprotonation occur at opposite sides of the planar, sp2-hybridized intermediate. In the epimerase reaction shown here, a coordinated pair of aspartate residues is located at opposite sides of the enzyme's active site. Asp1, which is ionized at the start of the catalytic cycle, abstracts the C3 proton from the front side of the plane of the page, converting C3 from a chiral tetrahedral center to an achiral, planar group. When reprotonation occurs, it is Asp2, on the back side of the plane of the page, which donates the proton, resulting in inversion of stereochemistry at C3 (J. Molec. Biol. 2003, 326, 127-135).
Notice another thing about this epimerization reaction: the key intermediate is an enolate anion, stabilized by coordination to a zinc cation (a Lewis acid). Contrast this with the phosphoglucase isomerase reaction (section 13.2A), where stabilization of the key intermediate was achieved by proton donation to form a short-lived enol species.
There are many more examples of reactions in which the stereochemistry of an a-carbon in a carbonyl compound is inverted. The short polypeptides that form part of the peptidoglycan cell wall structure in bacteria contain some D-amino acids, which have the opposite stereochemistry at the a-carbon compared to the L-amino acids that are found almost exclusively in proteins. Enzymes called amino acid racemases catalyze the conversion between the L and D forms of these amino acids. While many amino acid racemases depend on the coenzyme pyridoxal phosphate to stabilize the key carbanion intermediate (we'll study these reactions in section 14.4B), others proceed through an enolate intermediate. Glutamate racemase, which converts L-glutamate to D-glutamate, is an example (Biochemistry 2001, 40, 6199). Although many mechanistic details are as yet unknown, it has been demonstrated that, at least for some of the cofactor-independent racemases, a pair of cysteine residues serves as the catalytic acid/base pair.
Exercise 13.4: Propose a likely mechanism for glutamate racemase, showing stereochemistry throughout.
13.2C: Alkene isomerization in the degradation of unsaturated fatty acids
The oxidation pathway for unsaturated fatty acids (in other words, fatty acids whose hydrocarbon chains contain one or more double bonds) involves the 'shuffling' of the position of carbon-carbon double bonds in the chain. This is accomplished by enoyl CoA isomerase, through an enolate intermediate. Here, the cis double bond between C3 and C4 is isomerized to a trans double bond between C2 and C3.

The catalytic acid and base groups are glutamate and/or aspartate residues (J. Biol Chem 2001, 276, 13622).
In section 15.3A, we will see a different example of an alkene isomerization reaction mechanism in which the key intermediate is a carbocation, rather than an enolate.