
Formation of alcohols from alkenes
- Page ID
- 15411
Alkenes can be converted to alcohols by the net addition of water across the double bond. There are multiple ways that are commonly used to do this transformation.
Hydration of Alkenes
The net addition of water to alkenes is known as hydration.

The result involves breaking the pi bond in the alkene and an OH bond in water and the formation of a C-H bond and a C-OH bond. The reaction is typically exothermic by 10 - 15 kcal/mol,1 but has an entropy change of -35 - -40 cal/mol K. Consequently, the net free energy change for the process tends to close to 0, and the equilibrium constant for the direct addition is close to 1. Nonetheless, there are multiple approaches that allow this transformation to be carried out to completion.
Acid-Catalyzed Hydration
The direct addtion of water to an alkene is too slow to be of any significance. However, the addition can be catayzed by Lewis or Bronsted acids.

The mechanism of hydration involves electrophlic addition of the proton (or acid) to the double bond to form a carbocation intermediate. Addition of water in the second step results in formation of an oxonium ion, which, upon deprotonation, gives the alcohol.

The proton in the oxonium intermediate can be deprotonated by any base present, including the conjugate base of the acid used as a catalyst, or even by another alkene molecule, which would generate another carbocation intermediate and propagate the chain mechanism.
Using Le Châtelier's Principle to get the product
The extent of reaction of the acid catalyzed hydration of alkenes is determined by the equilibrium constant, which, as noted above, is near unity. Consequently, the reaction can be carried out by adding excess water to increase the yield of products, an application of Le Châtelier's Principle. Alternatively, the reverse reaction, acid-catalyzed dehydration of the alcohol to form the alkene can promoted by removing water from the reaction by using a Dean-Stark trap.
Regioselectivity of addition
Protonation of the alkene in the first step of the reaction can occur at either carbon. However, the more stable carbocation is preferably formed. The order of stability of alkyl cations is
3o > 2o > 1o
Therefore, protonation will occur at the less substituted carbon, to create the more substituted carbocation, where the water adds. The addition of a proton at the less substituted carbon and the -OH to the more substituted carbon is known as Markovnikov's Rule.
The carbocation formed in the reaction is prone to rearrangement, if possible. Therefore, hydration of an olefiin next to a branched aliphatic center will result in the alcohol forming in a position that was not part of the original double bond.

Oxymercuration
Transition metals can also be used as the acids for hydration reactions. In some cases, coordination of the alkene to a metal leaves it susceptible to reaction with a nucleophile such as water. The classic case of nucleophilic donation to a coordinated alkene occurs with mercury (II) salts such as mercuric chloride, \(HgCl_2\), or mercuric acetate, \(Hg(OAc)_2\). The reaction, or rather the sequence of reactions, is called oxymercuration - demercuration or oxymercuration - reduction.
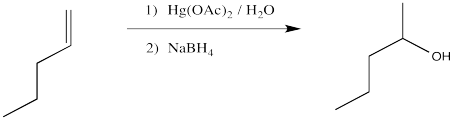
We will break the two different reactions in this sequence apart and focus only on the first one: oxymercuration. This reaction qualifies as an electrophilic addition because, as in the previous cases, it begins with donation of a π-bonding pair to an electrophile. In this case, we will consider the electrophile to be aqueous \(Hg^{2+}\) ion.
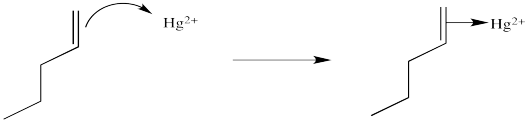
That electrophilic addition (from the alkene's perspective) results in the formation of an alkene complex. In reality, the mercury ion is also coordinated by several water molecules, but we will ignore them for simplicity.
The complex formed by addition of mercury is not a localized carbocation, as is formed by protonation, but is better considered a bridged or cyclic structure, which results from addition of d electrons to the empty orbital in the cation.
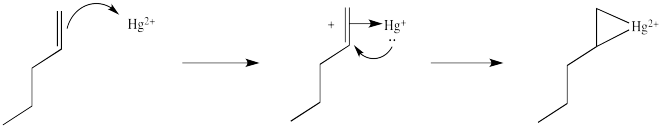
This situation is something like formation of a cyclic bromonium ion formed in the bromination of alkenes. As with bromination, the cyclic intermediate can be opened by attack of a nucleophile, in this case, water.

The final part of the reaction sequence is displacement of mercury from the hydroxyalkylmercury complex, effected through addition of sodium borohydride. The details of the reaction are usually dismissed in textbooks because they have little to do with electrophilic addition, the topic we are focusing on. However, the result is that the mercury is replaced by a hydrogen atom. The metal is converted to silvery, liquid, elemental mercury.

Regioselectivy of hydration by oxymercuration
As shown above, oxymercuration leads to Markovnikov addition of water to the double bond. The reason for this can be seen by considering the electronic structure of the cyclic cation. The electronic structure of the cyclic intermediate can be deduced by using resonance structures as shown below.
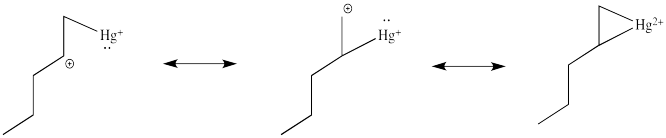
By considering these resonance structures, it can be seen that the postive charge is distributed over both of the carbons of the olefin. However, because the resonance structure on the left has the positive charge on a secondary carbon, it is energetically more favorable than the structure in the middle, where the charge is a primary cation. Consequently, although the charge is distributed over both carbons, the more substituted carbon has more positive charge density in the overall resonance hybrid. Therefore, it is more electrophilic, and is more susceptibleable to nucleophlic attack.
An important advantage of oxymercuration over simple acid catalysis is that the cyclic structure is not prone to rearrangement, and can therefore is amenable to hydration of alkenes with branched substituents.
