
Spontaneity
- Page ID
- 24708
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\dsum}{\displaystyle\sum\limits} \)
\( \newcommand{\dint}{\displaystyle\int\limits} \)
\( \newcommand{\dlim}{\displaystyle\lim\limits} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\(\newcommand{\longvect}{\overrightarrow}\)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)Before bringing in the final piece of the puzzle of spontaneity it would be a good idea to define this term we have been using so freely. In ordinary conversations "spontaneous" is generally taken to mean something like "happens by itself". This certainly does not seem to describe the thermite reaction which requires significant intervention to initiate.
That last word of the previous sentence is important. All reactions require some minimum amount of energy to begin. Some require so little that we never notice (precipitations, for example). Some--like the thermite reaction--require a lot, But that energy is not part of the spontaneity puzzle.
In simple terms a process is spontaneous if it proceeds on its own once initiated.
In more technical terms, a process is spontaneous if it transfers free energy from the system to the surroundings.
In 1875 J. Willard Gibbs defined the spontaneity of a reaction in terms of its ability to perform "useful work" (in principle or practice). This ability to do work is determined, according to Gibbs, by the change in free energy during a chemical change, ΔG. This quantity gives the maximum amount of work obtainable from a spontaneous reaction (OR it tells how much work must be done to make a reaction spontaneous).
The sign of ΔG in an indicator of whether a reaction will be spontaneous or not.
- when ΔG < 0, the reaction is spontaneous (it is capable of doing useful work)
- when ΔG = 0, the system is at equilibrium
- when ΔG > 0, the reaction is non-spontaneous (work must come from the surroundings to make the reaction happen)
Free energies of formation (ΔGfo) have been tabulated just like enthalpies of formation or absolute entropies and can be used in the same way to determine the overall free energy change in a process.
Examples
This still leaves open the question of temperature. All of these values are determined for "standard states" (1 atmosphere pressure, 1 M concentration) and 298 K. What if the temperature is different?
This is where life becomes "interesting". Recall that when we talked about the entropy change in the surroundings it mattered whether the temperature of the surroundings was low or high. It is time to take another look at the energy/entropy picture in order to sort this out.
Example
Consider the displacement of aqueous silver ions by copper metal:
\[Cu + 2 Ag^+ → Cu^{2+} + 2 Ag\]
ΔHorxn for this process is -147.2 kJ and \(ΔG^o_{rxn}\) is -88.3 kJ. Based on our established criteria, this process will be spontaneous at standard states. One way to look at these two energy terms is this:
- ΔH represents the total amount of energy that must be transferred to the surroundings in order to keep the temperature of the system at 298 K
- ΔG represents the part of that total energy which can be harnessed to do useful work during the transfer, i.e., the energy "free" to do work
From this we might say:
ΔH = ΔG + "unfree" energy
Or, put in terms of what we can literally expect in useful energy output from a process:
ΔG = ΔH - "unfree" energy
Experiments show that the amount of free energy obtainable from a process is dependent on temperature and since enthalpy values change only a little with temperature, the "unfree" energy term must include a temperature dependence.
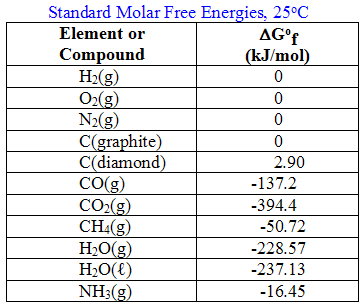
By now you might suspect--and correctly--that the "unfree" energy term involves what we have called the entropy change, ΔS. We know that the entropy change of the system is connected to the entropy change of the surroundings which is highly temperature dependent. Thus the relationship above is generally written as:
ΔGo = ΔHo - TΔSo
This is known as the Gibbs-Helmholtz equation and is the absolute bottom line in chemical thermodynamics. Sort of. So we are finally ready to take on temperature....
Calcium carbonate occurs in a number of natural forms, limestone being one of the most plentiful. If heated sufficiently, the calcium carbonate decomposes into calcium oxide (quicklime), which is used by the cement industry, and carbon dioxide:
\[CaCO_3 \rightarrow CaO + CO_2\]
To determine if this process is spontaneous at standard states and 298 K we have a couple of options:
- calculate ΔGorxn from ΔGof values
- calculate ΔGorxn from ΔHorxn and ΔSorxn
The first option seems easier but the second one is more useful as you will see.

O.K. So we are not going to get rich any time soon trying to produce CaO at standard conditions. So can we do it at all? Is there a temperature at which the process will become spontaneous?
As a thought-problem, this one is fairly simple. The entropy in the system increases. This is good. The entropy in the surroundings decreases since heat is transferred to the system in this endothermic process. How can we make that loss in entropy in the surroundings less significant?

Raising the temperature should offset the decrease in entropy in the surroundings. But how high? We don't want to waste money on a lot of fuel we don't need.
This kind of calculation is an approximation since we are using the standard enthalpy and entropy values to calculate at a non-standard temperature. This is justified for two reasons.
- both ΔHo and ΔSo change only a little with temperature
- both ΔHo and ΔSo change in the same direction so the effect tends to cancel somewhat during subtraction
This general method is limited to standard atmospheric pressure and 1 M concentrations (for ions in solution), however. A further treatment is beyond our needs at this level.
Some additional thought about the Gibbs-Helmholtz equation will reveal that the relationship it describes can be used to predict those reactions which can be manipulated by changing temperature.
A decision on the spontaneity of a reaction using ΔGo is frequently not an either/or matter. We say that a process is "non-spontaneous" if ΔGo > 0 but we can see from the previous example that temperature might change that situation.
In fact, the possibilities can be roughly divided into four parts:
- ΔGo < -60 kJ: reaction "complete"
- -60 kJ < ΔGo < 0 kJ: mostly products
- 0 kJ < ΔGo < +60 kJ: mostly reactants
- ΔGo > +60 kJ: "no" reaction
These various cases imply that chemical processes are reversible and that is theoretically true. In practice, many are not. The manipulation of the extent of reaction or making a non-spontaneous process happen can be visualized as four scenarios based on the Gibbs-Helmholtz relationship. These ideas can be applied to reactions on the industrial scale (the cement again...) as well as to mundane matter such as when water will boil.

So free energy finally gives us a tool for predicting whether a reaction will occur or not under specific conditions. Or does it?

Sometimes things just don't work out.

That is the case for thermodynamics as THE predictive tool for chemical reactions. If in fact thermodynamic considerations were the only matters of consequence in determining whether a reaction should work, we would all be in the lab right now, making more diamonds than you could shake a stirring rod at.
So we've reached the end of the beginning. And we've answered two of the four big questions.
