
GAS-PHASE ION THERMOCHEMISTRY
- Page ID
- 14854
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\dsum}{\displaystyle\sum\limits} \)
\( \newcommand{\dint}{\displaystyle\int\limits} \)
\( \newcommand{\dlim}{\displaystyle\lim\limits} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\(\newcommand{\longvect}{\overrightarrow}\)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)Sharon G. Lias and John E. Bartmess
Summary
The WebBook includes a database concerned with ion thermochemistry that presents a collection of experimentally-determined values for
of molecules or molecular fragments. In addition, data for proton affinities, and gas phase basicities from a new re-evaluation of the thermochemical scales is included. Guidance is provided (at least for the more common molecules) in the selection of a "best" value, when several different values have been reported for a quantity. Values for the gas phase enthalpies of formation of some of the common positive and negative ions derived from these data are displayed. The values for enthalpies of formation are presented using the so-called Ion Convention for treating the thermochemistry of the electron (rather than the Electron Convention commonly used in some thermodynamics compilations).
In the accompanying text, definitions of all the above-listed quantities are provided. The coverage of data in the collection is outlined, and explained in terms of the history of the database. The units used are explained, and a tutorial discussion of thermochemical conventions for the electron is provided, including instructions for converting data from one convention to the other, and a value for the integrated heat capacity of the electron calculated using the correct Fermi-Dirac statistics. Scientific background necessary to an understanding of the interpretation of these kinds of data is presented, and includes descriptions of the Franck-Condon Principle, the meanings of the terms "hot bands" and "kinetic shift", as well as the role of potential barriers in the reaction coordinate in the interpretation of appearance energy data. The derivation of gas phase thermochemical information on ions from the interpretation of Rydberg series and from ion/molecule equilibrium constant determinations is also discussed.
A description of the methods from which the various kinds of data are derived is provided. This includes descriptions of the experimental techniques:
- electron ionization techniques
- photoionization mass spectrometry
- time-resolved photodissociation
- laser spectroscopy
- photoelectron spectroscopy
- threshold electron detection
- photoion-photoelectron coincidence spectroscopy
- chemi-ionization
- charge exchange mass spectrometry
- neutral beam ionization/appearance energy
- electric field detachment
In addition, the derivation of thermochemical information from ion/molecule reaction studies (including ion/molecule equilibrium constant determinations, determinations of reaction endothermicity, ion/molecule bracketing experiments, and the kinetic branching method are discussed. Descriptions are also included of techniques used less often for the derivation of ion energetics information, for example:
- charge inversion energy loss spectroscopy
- collisionally-activated dissociation
- delayed thermionic ionization
- charge transfer spectra
- Auger electron spectroscopy
- surface ionization
- liquid phase electrochemical determinations
- electron auto-detachment rate
- electron transmission spectroscopy
- kinetic mobility of ions
- laser optogalvanic spectroscopy
- zero-pressure thermal radiation induced dissociation
- scattering of atoms from ions
The use of derivations, estimations, literature evaluations, theoretical calculations, and lattice energy calculations, in the collection is also described.
A short discussion of the reliability of data and criteria for evaluation presents comparisons among results of different techniques and a description of the handling of error limits in the database.
Because data on positive ions and negative ions, derived from separately maintained databases, are integrated here for the first time, the WebBook user may notice that there are some inconsistencies in the way analogous items are treated; a description of these inconsistencies is provided to aid the WebBook user in searching for these items.
Outline
- Introduction
- Definitions
- Ionization Energy
- Appearance Energy
- Proton Affinity and Gas Phase Basicity
- Electron Affinity
- Gas Phase Acidity and Relative Acidity
- Enthalpies of Formation of Ions in the Gas Phase
- Thermochemical Conventions for Enthalpies of Formation of Ions
- History, Coverage, and Presentation of the Database
- Units
- Thermochemical Conventions for the Electron
- Definitions
- Interpretation of Ion Thermochemistry Data
- Experimental Techniques
- Optical Spectroscopy [S]
- Threshold Experiments
- Electron Ionization Techniques [EI]
- Photoionization Techniques
- Photoionization Mass Spectrometry [PI],[PD],[LPD]
- Time-Resolved Photodissociation [TRPI]
- Laser Spectroscopy [LS] (multi-photon ionization and resonance ionization mass spectrometry)
- Photoelectron Spectroscopy [PE],[LPES]
- Threshold Electron Detection [TE],[ZEKE]
- Photoion-Photoelectron Coincidence Spectroscopy [PIPECO]
- Chemi-Ionization [CI]
- Charge Exchange Mass Spectrometry [CEMS]
- Neutral Beam Ionization/Appearance Energy [NBIE],[NBAE]
- Electric Field Detachment [EFD]
- Thermochemical Information from Studies of Ion/Molecule Reactions
- Other Experimental Techniques
- Charge Inversion Energy Loss Spectrometry [CIEL]
- Collisionally-Activated Dissociation [CAD],[CIDT]
- Delayed Thermionic Ionization [DTI]
- Charge Transfer Spectra [CTS]
- Auger Electron Spectroscopy [AUG]
- Surface Ionization [SI]
- Liquid-Phase Electrochemical Determinations [LE]
- Electron Auto-Detachment Rate [KINT]
- Electron Transmission Spectroscopy [ETS]
- Kinetic Mobility of Ions [Mobl]
- Laser Optogalvanic Spectroscopy [LOG]
- Zero-pressure Thermal radiation Induced Dissociation [ZTID]
- Scattering of Atoms from Ions [Scat]
- Non-Experimental Methods
- Reliability of Data and Criteria for Evaluation
- References
INTRODUCTION
The database concerned with ion thermochemistry included in the WebBook makes available a reasonably complete listing of values for
- ionization energies
- appearance energies
- electron affinities
- acidities
- proton affinities and gas phase basicities
of molecules or molecular fragments. Data displayed for ionization energies, electron affinities and acidities are experimental values as reported in the literature, and are given along with a citation of the original paper; values for proton affinities and gas phase basicities are taken from a new re-evaluation of the relevant thermochemical scales, and original references are not given in the current version of the WebBook. For the experimental data, guidance is provided (at least for the more common molecules) in the selection of a "best" value, when several different values have been reported for a quantity. Values for the gas phase enthalpies of formation of some of the common positive and negative ions derived from these data are displayed. The values for enthalpies of formation are presented using the so-called Ion Convention for treating the thermochemistry of the electron (rather than the Electron Convention commonly used in thermodynamics compilations).
In the August 1997 release of the WebBook, for the first time, the NIST Positive Ion (ionization energies, appearance energies), Proton Affinity (gas phase basicities, proton affinities) and Negative Ion (electron affinities, gas phase acidities) databases are combined into a single searchable Ion Energetics database. Because the Positive and Negative Ion databases were developed and maintained separately, the structures of the two sets of files are different, resulting in some inconsistencies in the way analogous data are presented in the WebBook. In future updates, these inconsistencies will be removed, but at the present time users should be aware that (a) data relevant to formation of positive ions are accessed through a search for the precursor neutral species while data on the thermochemistry of the anion is found by searching for the anion; (b) when the box on the entry screen denoting a search for "Ion Energetics" data is checked, available data on gas phase basicities and proton affinities will be displayed with every "hit", while available data on gas phase acidities will not appear; in order to access gas phase acidity data, the user must also click on the box in the entry screen labeled "Reaction". For the same reason, the user will find that sometimes the same experimental technique is designated by two different acronyms in accessing results on positive and negative ions; because these acronyms are fully defined, it is hoped that this will not cause confusion.
The current collection is an update and extension of several earlier data compilations / evaluations, including all the data which appeared in previous publications [1, 2, 3, 4, 7] and electronic databases [5, 9], and the new re-evaluation of the proton affinity scale [10], as well as data which has appeared in the literature since the release of the most recent electronic database [9].
Definitions
Ionization Energy
The ionization energy, sometimes called (less correctly) the ionization potential (usually designated by IE, IP, or, in the older literature, I), is the energy required to remove an electron from a molecule or atom:
M → M+ + e− ΔHrxn= IEa
Ionization energies are characterized as adiabatic or vertical values.
Adiabatic Ionization Energy
The adiabatic ionization energy is the lowest energy required to effect the removal of an electron from a molecule or atom, and corresponds to the transition from the lowest electronic, vibrational and rotational level of the isolated molecule to the lowest electronic, vibrational and rotational level of the isolated ion. Adiabatic ionization energy data can be used to obtain values for the enthalpy of formation of the ion, M+.
Vertical Ionization Energy
The vertical ionization energy is the energy change corresponding to an ionization reaction leading to formation of the ion in a configuration which is the same as that of the equilibrium geometry of the ground state neutral molecule. According to the Franck-Condon principle, when a molecule is ionized by photoionization or by interaction with energetic electrons, the highest probability configuration of the resulting ion will be the configuration of the precursor neutral molecule. When the equilibrium geometry of the ion is very similar to that of the neutral precursor molecule, the vertical and adiabatic ionization energies will be the same, or nearly so. The vertical ionization energy must always be greater than or equal to the adiabatic ionization energy.
Appearance Energy
Since ionization energies are often determined in experiments in which the ionizing photon or electron energy is varied until the appearance of an ion is observed ("threshold measurements"), ionization energies have sometimes been called appearance energies. However, in general usage this term has come to have a more specific meaning. As used here, and in most of the technical literature, the term appearance energy, sometimes (less correctly) called the appearance potential, refers to the minimum energy required to form a particular fragment ion from a precursor neutral molecule:
AB → A+ + B + e− Δ Hrxn = AP
For more information see the description of the presentation of appearance energy data, the discussion of the use of appearance energy data to derive enthalpies of formation of fragment ions, as well as the description of some of the complicating factors involved in the interpretation of appearance energy data.
Proton Affinity and Gas Phase Basicity
Stable cations formed in the gas phase also include protonated neutral molecules, generated in proton transfer reactions. Formally, the relationship between the enthalpy of formation of MH+ and its neutral counterpart, M, is defined in terms of a quantity called the proton affinity, PA. This the negative of the enthalpy change of the hypothetical protonation reaction:
M + H+ → MH+ Δ Hrxn = −PA
ΔfH;(MH+) = ΔfH°(M) + ΔfH;(H+) − PA
The term proton affinity, as universally used, is a quantity defined at a finite temperature, usually 298 K, and is therefore not strictly analogous to the adiabatic ionization energy or electron affinity, both of which are the 0 K enthalpy changes of the corresponding reactions.
At 298 K, the enthalpy of formation of the proton, using the Ion Convention (or "stationary electron" convention), is 365.7 kcal/mol, 1530.0 kJ/mol.
The Gibbs energy change associated with the protonation reaction is called the gas phase basicity, GB, of molecule M. Most available data on gas phase basicities and proton affinities has been obtained from experiments in which the equilibrium constants of proton transfer reactions are determined. See the discussion of derivation of thermochemical data from ion/molecule equilibrium constants for relevant equations and details.
Electron Affinity
The electron affinity, EA, of a molecule is, for negative ions or anions, the quantity that is analogous to the ionization energy for positive ions. That is, the electron affinity is equal to the energy difference between the enthalpy of formation of a neutral species and the enthalpy of formation of the negative ion of the same structure. The electron affinity is defined as the negative of the 0 K enthalpy change for the electron attachment reaction:
M + e− → M− Δ Hrxn = −EAa
-EA = ΔfH;(M−) − ΔfH°(M) − ΔfH;(e−)
As with the ionization energy, it is possible to have either vertical or adiabatic electron affinities, with the numeric value of the vertical quantity being greater than or equal to the adiabatic value. A difference from the ionization energy is that for stable ("bound") negative ions, the ion is lower in energy than the corresponding neutral. If the negative ion is higher in energy than the neutral, the neutral is said to have a negative electron affinity, and the ion will usually undergo spontaneous loss of the electron.
Gas Phase Acidity and Relative Acidity
The gas phase acidity (or merely, acidity) of a molecule AH, Δacid G(AH), is the Gibbs energy change of the reaction:
AH → A− + H+
usually defined at 298 K. The enthalpy change of this reaction, Δacid H, is, of course, the proton affinity of the anion. The Gibbs energy change of the reaction:
AH + B− → BH + A−
is called the relative acidity of species AH and BH. Most data are derived from determinations of the equilibrium constant of this reaction. See the discussion of derivation of thermochemical data from ion/molecule equilibrium constants for relevant equations and details.
Enthalpies of Formation of Ions in the Gas Phase
The enthalpy of formation (or heat of formation) of an ion in the gas phase can, in principle, be obtained through a straightforward treatment of the thermochemistry of the ionization process. For example, the enthalpy of formation of a positive molecular ion is obtained by adding the enthalpy of formation of the precursor molecule to the adiabatic ionization energy (IE) and subtracting the enthalpy of formation of the electron,
ΔfH;(M+) = ΔfH°(M) + IEa − ΔfH;(e−)
while that of an anion is based on the analogous quantities combined with the value for the electron affinity:
ΔfH;(M−) = ΔfH°(M) − EA + ΔfH;(e−)
Similarly, enthalpies of formation of positive fragment ions, A+, are given by:
ΔfH;(A+) = ΔfH°(AB) − ΔfH°(B) − ΔfH;(e−) + AP
assuming that there is no potential barrier in the reaction coordinate for the dissociation reaction, and little or no kinetic shift.
Note that in the equations given here, there is no reference to temperature, but in practice, one may have to be concerned about the fact that the ionization energy or electron affinity is a quantity corresponding to a zero degrees Kelvin process, while the available data on the enthalpy of formation of the neutral species may correspond to some higher temperature. This presents a complication (especially for those interested in highly accurate data) in the derivation of enthalpies of formation of ions in the gas phase. A rigorous treatment of enthalpies of formation of ions at finite temperatures requires a consideration of the changes in the ionization energy/enthalpy of formation with temperature. However, when high accuracy is not required, the simplifying assumption that the adiabatic ionization energy is approximately equivalent to the 298 K enthalpy of ionization is usually adequate. (For a more detailed presentation, see the discussion of the thermochemistry of ions in the gas phase at finite temperatures.)
In this compilation, the Ion Convention for treating the thermochemistry of the electron is used. This essentially means that the terms involving the electron can be ignored in these equations.
Thermochemical Conventions for Enthalpies of Formation of Ions
In order to derive enthalpies of formation of positive or negative ions in the gas phase using ionization or appearance energy data, it is necessary to treat the thermochemistry of the electron. There are two conventions, both in widespread use, for dealing with the thermochemistry of the electron, one (the Ion Convention) used by the ion chemistry/physics community, and one (the Electron Convention) used by thermodynamicists.
The Ion Convention
According to the "Ion Convention" (sometimes unfortunately called the "stationary electron convention") which is adopted here, the enthalpy of formation of the electron at non-zero temperatures is equal to the integrated heat capacity of the electron; when data are treated this way (for details, see discussion on Thermochemical Conventions for the Electron), the enthalpy of formation of the electron cancels out in calculating the enthalpy of formation of an ion. That is, in this convention, which is adopted by most mass spectrometrists and other ion physicists and chemists, the thermochemistry of the electron can be ignored when deriving an ion enthalpy of formation.
The Electron Convention
Thermodynamicists commonly use the "Electron Convention" which treats the electron as a standard chemical element with an enthalpy of formation defined as zero at all temperatures. Since standard thermodynamics is a non-rationalized system, the effect of constraining the enthalpy of formation of the electron gas to zero at all temperatures, is that the electron's integrated heat capacity must be accommodated elsewhere in the equation — namely, in the derived value for the enthalpy of formation of the ion. Therefore, in standard thermodynamics data compilations [6], cited values for enthalpies of formation of ions at temperatures other than 0K differ from those given here. Because in standard thermodynamic data compilations [6], the integrated heat capacity of an electron gas has commonly been taken to be the same as that of an ideal gas following Boltzmann statistics − 5/2 RT — a value for an "Ion Convention" enthalpy of formation of a positive ion is numerically more negative than the value in the "Electron Convention" by 5/2 RT (6.2 kJ/mol at 298 K); that for a negative ion, is more positive by the same amount.
History, Coverage, and Presentation of the Database
History
The collection of gas phase ion energetics data presented in the WebBook traces its origin to a series of publications concerned with this subject area, beginning with a table of ionization energies and evaluated enthalpies of formation of ions included in the 1957 book "Electron Impact Phenomena and the Properties of Gaseous Ions" by F. H. Field and J. L. Franklin [1]. In 1969, H. M. Rosenstock and collaborators, from the National Bureau of Standards, joined Field and Franklin to expand that table in the publication "Ionization Potentials, Appearance Potentials, and Heats of Formation of Gaseous Positive Ions" [2]. In 1977, H.M. Rosenstock, K. Draxl, B.W. Steiner, and J.T.Herron published an update, "Energetics of Gaseous Ions," [3] which included a complete re-evaluation of the data, and for the first time, a table of electron affinity data.
In 1982, an extensive compilation of ionization potential and appearance potential data, "Ionization Potential and Appearance Potential Measurements, 1971-1981" [4], presented unevaluated measurements which had appeared in the literature from the 1971 cut-off date of the previous collection [3] up to mid-1981. In 1988, an evaluation or re-evaluation of the data from the collective database presented in all these earlier publications [1, 2, 3, 4], was published in "Gas-phase Ion and Neutral Thermochemistry" [7] (commonly referred to as "the GIANT Tables"). The 1988 publication also included more recent data and evaluated proton affinity values, as well as a comprehensive table of data on negative ions, including both electron affinity and gas phase acidity values. The data on proton affinities were taken from a 1984 publication [8] in which the entire thermochemical scale of gas phase basicities and proton affinities had been evaluated.
The 1988 publication of evaluated data on ion thermochemistry [9] was made available by the National Institute of Standards and Technology's Standard Reference Data program as a searchable computer database available on diskettes — in fact, as two jointly-distributed databases, one presenting the data relevant to positive ions [9a] and the other, data on negative ions [9b]. In the initial release, the negative ion database [9b] displayed the entire corpus of data from which the evaluations were drawn, but the original version of the electronic database on positive ions [9a] presented only evaluated values for ionization energies, proton affinities, and enthalpies of formation of positive ions in the gas phase. The 1991 and 1993 updates [5] to the electronic database added more recent data, and, in the case of the positive ion collection [5a], for the first time displayed some of the original data (e.g. data published after 1971) from which the evaluations were drawn.
After new experimental determinations had made necessary a re-evaluation of the entire proton affinity scale as presented in the 1984 publication [8] (data derived from equilibrium constant determinations are interdependent and must be evaluated collectively), proton affinity data which had appeared in the original electronic database [9a] were removed from the updated version [5] to prevent dissemination of out-of-date information. The re-evaluation of the proton affinity scale has now been completed [10], and the evaluated proton affinity and gas basicity data are included in the WebBook.
Since most of these precursor publications are still in widespread use (even the woefully out-of-date 1969 compilation [2] continues to be cited), it is worthwhile to summarize the contents and coverage of the various publications, and compare these to the contents and coverage of the WebBook:
Summary
- Field and Franklin, 1957 [1]
- Table of ionization energies and enthalpies of formation of positive ions
- Franklin et al, 1969 [2]
- Comprehensive collection of ionization/appearance energy measurements with evaluations where possible and enthalpies of formation of some positive ions; cut-off date, mid-1966.
- Rosenstock et al, 1977 [3]
- Comprehensive collection of ionization/appearance energy and electron affinity measurements with evaluations where possible and enthalpies of formation of some positive and negative ions; cut-off date, mid-1971.
- Levin and Lias, 1982 [4]
- Comprehensive collection of ionization/appearance energy data; no evaluations; covers literature from mid-1971 to mid-1981.
- Lias, Liebman and Levin, 1984 [8]
- Evaluation of the scale of gas phase basicities/proton affinities with data for 820 molecules.
- GIANT Tables, 1988 [7]
- Positive ions: Evaluated ionization energy values, proton affinities, and enthalpies of formation; original measurements and appearance energies not displayed.
Negative ions: Comprehensive collection of electron affinity and gas phase acidity data with evaluations and anion enthalpies of formation.
Cut-off date for literature coverage, mid-1986. - GIANT Tables PC Version 1.0, 1990 [9]
- Same as 1988 GIANT Tables
- GIANT Tables PC Versions 2.0/3.0, 1991-93 [5]
- Original ionization energy measurements 1971-1992 displayed along with evaluations from 1988 GIANT Tables. Proton affinity data removed. Electron affinity and gas phase acidity data updated.
- NIST WebBook, Feb. 1997
- Only positive ion data provided; original ionization energy measurements from all earlier references displayed, with evaluations from 1988 GIANT Tables. Newly-evaluated proton affinity data added. Literature update through mid-1996.
- NIST WebBook, August 1997
- Addition of updated negative ion data (electron affinities, gas phase acidities, and cluster ion data). Some new ionization energy/appearance energy data added, and some evaluations of positive ion data revised.
- Hunter and Lias, 1998 [10]
- Re-evaluation of the gas phase basicity/proton affinity scale with data for 1740 molecules.
Coverage
The ion energetics database at the present time has as its focus numeric data concerned with the energies for formation of particular positive and negative ions in the gas phase, as well as with the energetics of certain reactions, namely protonation and deprotonation, of those ions. That is, the database includes values for ionization energies, appearance energies, electron affinities, gas phase acidities and basicities, as well as proton affinities. In addition, the database contains data on the energies associated with the clustering of neutral molecules to anions.
Thermochemical information about positively-charged ion/molecule clusters has been compiled, but has not yet been incorporated into the WebBook database. Some information on ionization energies of small cluster ions (with not more than three or four ligand molecules), especially in inorganic systems, is included. The database does not at the present time include thermochemical data derived from collisionally-activated dissociation experiments of positive ions, although such results for negative ions are included.
In recent years, numerous publications giving quantum mechanical calculations of very high accuracy on the thermochemical properties of ions, especially small ions, have appeared. The present work includes only data derived from experimental determinations, although some calculational results have been used in carrying out evaluations, especially of the proton affinity data.
A particular explanation about the coverage of ionization energy data is in order. In most of the precursor publications [1, 2, 3, 4, 7] the primary focus was the thermochemistry of ions in the gas phase. That is, the goal of collecting ionization energies, appearance energies, electron affinities, and so forth was the derivation of enthalpies of formation of ions. Because of this emphasis, the coverage of data was restricted to information directly relevant to deriving ionic enthalpies of formation. This limited focus had particular implications for the coverage of data on ionization energies, since it necessarily meant that only data on the lowest ionization energies was included; ionization energies leading to the formation of excited ions, or multiply charged ions were excluded (except for atoms and diatomic molecules in the 1977 publication [3]). Furthermore, coverage was restricted to adiabatic ionization energy data except in cases where publications gave only vertical ionization energy values. In abstracting data from the more recent literature (since 1993), we have attempted to include both adiabatic and vertical values where both are available, but have not gone back to the thousands of earlier papers to re-abstract data on vertical ionization energies or upper ionization energies that were not originally included.
Presentation
The positive ion database (ionization energies, appearance energies, proton affinities) and negative ion database (electron affinities, acidities) were developed and are maintained separately. As a result, certain inconsistencies exist at the present time in the presentation of the two types of data in the Web Book. For example, the primary sort in the anion database depends on the identity of the anion, while the positive ion database has always been organized around the identity of the precursor neutral molecule. For this reason, until further work has been accomplished, users of the WebBook will occasionally see an apparently illogical list of "hits" when data for a particular chemical species has been requested. It is intended that such inconsistencies be cleaned up up in the next version of the WebBook.
For example, when carrying out a sort for "ion energetics" data, if the database contains both ionization/appearance energy data and electron affinity data for species M, then M will be listed twice in the list of "hits", once as M (giving data on the energies associated with ionization of M to M+) and once as M- (with data on the electron affinity of molecule M). Retrieval of data listed under M will also lead to a table giving the proton affinity and gas phase basicity of molecule M, if they are available. Unlike gas phase basicity data, data on gas phase acidities can be retrieved only if you started the search by requesting data on "reactions" (in addition to "ion energetics") on the opening screen. The gas phase basicity/proton affinity data are, at the present time, presented in less detail.
The formal organization of the positive and negative ion data based on neutral precursor (positive ions) or on the ion also has implications for carrying out a search for data based on registry number. For positive ion data, searches should be conducted based on the registry number of the neutral species; for anion data searches should be conducted on the registry number of the anion. Since the database contains registry number data for a limited portion of the anions in the database, a chemical formula search will often be a better choice for finding a specific anion.
Appearance energy data are presented in two ways. First, by calling up all data for a particular molecule, you access a listing of all ionization energy determinations, and also the appearance energies that have been determined for fragmentation processes of that parent ion. Second, if you are interested in knowing the appearance energies determined for the formation of a particular ion through fragmentation of larger species, you are given the option (at the top of the display) to "View reactions leading to ____+ (ion structure unspecified)". By clicking on this text, you will access a list of all appearance energies determined for formation of fragment ions of that particular formula from other (larger) molecular species. It should be understood that in the great majority of cases, the structure of the fragment ion is not specified, and such data should be used mainly as auxiliary information, or as a guide to the original literature. However, in some cases where the original authors have carried out a sufficient analysis to be able to specify the structure of the fragment ion, this structure is indicated. (In other cases, the structure may be obvious, and such an indication is not necessary.) A word of caution is in order: again, in earlier versions of the database, such specifications were not included, and we have not yet gone back to fill in this missing information.
Units
Units used for the display of information are dictated by the current practices for reporting data of a particular kind. For example, ionization energy and electron affinity values are usually reported in electron volts, and that is the unit used here for these data. The user is given the option of displaying values for enthalpies of formation of ions, proton affinities, and gas phase basicities in kJ/mol or kcal/mol, both of which are widely used in the relevant literature.
The conversion factors which were used are: 1 electron volt (eV) = 23.06054 kcal/mol = 96.4853 kJ/mol; 1 kcal/mol = 4.184 kJ/mol, as given by the 1986 CODATA compilation [11].
Thermochemical Conventions for the Electron
The enthalpy of formation of any chemical species is defined as the difference between the enthalpy of the compound and the sum of the enthalpies of the elements of which it is composed. However, in the case of an ion, M+ or M−, one must explicitly take into account the enthalpy of the electron in some way. There are two widely-used conventions for dealing with the thermochemistry of the electron, one — called the "Electron Convention" — used predominantly by thermodynamicists and one — "the Ion Convention" — commonly adopted by scientists studying ion physics/chemistry. The "Ion Convention" is used here.
There is considerable confusion and misunderstanding of the basic assumptions and treatment of the thermochemistry of the electron in the two approaches. In fact, the so-called "electron" and "ion" conventions are really names assigned by the ion chemistry community to the two different conventions used by thermodynamicists for handling the integrated heat capacity of elements. The more widely-used convention (corresponding to the "electron convention") defines the enthalpies of formation of elements in their standard states to be zero at all temperatures; in this case, the integrated heat capacity of the element must be accommodated elsewhere in any thermodynamic equation, as shown in the more detailed discussion below — and in the particular case in which we are interested (ionization) end up as an increment in the derived enthalpy of formation of the ion. There is also a community of scientists in Europe (SGTE) who define the enthalpy of formation of elements at temperatures above absolute zero to be equal to the integrated heat capacity of the species; this corresponds to the treatment used for the electron in the so-called "ion convention". As will be derived below, when this treatment is followed, the integrated heat capacity term cancels out in the expression defining the enthalpy of formation of the ion. Of course, enthalpies of reaction are identical when using the two conventions (provided the same value is chosen for the integrated heat capacity term in question); the value of the integrated heat capacity merely appears on opposite sides of the equation, with opposite signs.
Further confusion exists because in the past, users of the two conventions have cited different values for the integrated heat capacity of the electron. Standard thermodynamics works [6], usually using the "electron convention", have taken a value of 5/2 RT (6.197 kJ/mol at 298 K), that is, the heat capacity of an ideal gas under Boltzmann statistics. Ion chemists/physicists, in deriving values for enthalpies of formation of ions, have in much of the earlier literature [7, 13] stated that they were assuming a value of zero for the integrated heat capacity of the electron; since the term cancels out in deriving an ion heat of formation in the "ion convention", the value chosen was, in fact, moot. A correct treatment of the heat capacity of an electron gas uses Fermi-Dirac, rather than Boltzmann, statistics [12]. A 1994 publication [12] arrives at a value of 3.145 kJ/mol (0.0326 eV) for the integrated heat capacity of an electron gas at 298 K, and is recommended for use in instances (such as reactions where the electron appears as a reactant) where the term does not cancel out.
Because of these differences in the treatment of the thermochemistry of the electron, values for the gas phase enthalpies of formation of ions at temperatures other than absolute zero cited in standard thermochemical compilations [6] differ from those given here or in most mass spectrometric literature, usually by 5/2 RT (6.197 kJ/mol at 298 K). Values for enthalpies of formation under the Electron Convention are higher (more positive) for positive ions and lower (less positive) for negative ions than the corresponding values expressed in the Ion Convention (used here). Problems arise when users unknowingly mix inconsistent values for enthalpies of formation in the same equation. The Table lists several commonly-used compilations and shows which convention is used in each.
Summary of Conventions Used for Electron Thermochemistry in Data Compilations
| Compilation | Convention | HT − H0 Included? c | ∫ Cp(e−) (kJ/mol) | ||
|---|---|---|---|---|---|
| M | M+/M− | e− | |||
| JANAF Tables [6a, 6b, 6c] | Electron | Yesa | Yesa | Yes | 6.197 |
| Gurvich et al. [6e] | Electron | Yesa | Yesa | Yes | 6.197 |
| NBS Tables [6d] | Electron | No | No | Yes | 6.197 |
| Rosenstock et al. [3] | Ion | Yesa | Yesa | N/Ab | N/Ab |
| GIANT [7] | Ion | Yesa | Yesa | N/Ab | N/Ab |
| NIST Database 19A Version 1.09 | Ion | Yesa | Yesa | N/Ab | N/Ab |
| NIST Database 19A Versions 2.0/3.05 | Ion | No | No | N/Ab | N/Ab |
| NIST Database 19B [5b, 9b] | Ion | Yesa | Yesa | N/Ab | N/Ab |
| WebBook | Ion | Yesa | Yesa | N/Ab | N/Ab |
a. When sufficient information is available.
b. N/A: Not applicable. See discussion.
c. Indicates treatment of integrated heat capacity terms for molecules and ions.
The following more detailed discussion of these issues is intended to present the question of how the electron is treated in a thermochemical equation in a tutorial manner, in the hope that some of the confusion will be dispelled. This discussion is also intended to justify the choice of the usual mass spectrometrists' convention for use in these tables.
The relationships between the various quantities that must be considered are shown in the thermochemical cycles:
| M0K | → | M+0K | + | e−0K | ΔHrxn = IEa |
| A↓ | B↓ | C↓ | |||
| M298 K | → | M+298 K | + | e−298 K | ΔHrxn = ΔHI |
and
| M0K | + | e−0K | → | M−0K | ΔHrxn = −EA |
| A↓ | C↓ | D↓ | |||
| M298 K | + | e−298 K | → | M−298 K | ΔHrxn = ΔHEA |
where A, B, C, and D are the integrated heat capacities for the various indicated species (e.g., A is the energy required to raise M from 0 K to 298 K), IEa and EA are the adiabatic ionization energy and the electron affinity (defined as the negative of the enthalpy change), and ΔHI and ΔHEA are the 298 K enthalpies of reaction. This discussion will be concerned with the standard temperature, 298 K, but the arguments can obviously be extended to any other temperature.
In both conventions, at 0 K the enthalpy of formation of the electron is zero and the enthalpies of formation of the ions are exactly equal to the 0 K enthalpy of formation of the molecule M plus the energy difference between M and the corresponding ion:
ΔfH;(M+)0K = ΔfH°(M)0K + IEa
ΔfH;(M−)0K = ΔfH°(M)0K − EA
At absolute zero, there is no difference in values derived using the two conventions.
When the temperature is raised to 298 K, the enthalpies of formation of M+ and M− will be related to the enthalpy of formation of M at 298 K through the enthalpy changes of the reactions at 298 K:
ΔfH(M+)298 K = ΔfH°(M)298 K − ΔfH;(e−)298 K + ΔHI
ΔfH(M−)298 K = ΔfH°(M)298 K + ΔfH;(e−)298 K + ΔHEA
The enthalpy changes of reaction at 298 K are related to the 0 K ionization energy and electron affinity through the relationships:
ΔHI = IEa + (C + B − A)
ΔHEA = − EA − (C + A − D)
In many treatments of thermodynamics of ions, the integrated heat capacity terms for M and the corresponding ion, M+ or M−, are taken to be approximately equal, i.e. A = B = D. In the present discussion, merely for the sake of focusing our attention on the treatment of C, the integrated heat capacity of the electron, let us temporarily make this assumption in order to simplify the equations. The validity of the usually-made assumption that A = B = D is considered in more detail in the section on thermochemistry of ions at finite temperature:
ΔfH;(M+)298 K = ΔfH°(M)298 K − ΔfH;(e−)298 K + [IEa + C]
ΔfH;(M−)298 K = ΔfH°(M)298 K + ΔfH;(e−)298 K − [EA + C]
In the Electron Convention, the electron is treated like a standard chemical element. Therefore, using the normal procedure for treating the thermochemistry of an element, its enthalpy of formation is constrained to be zero at all temperatures, but the integrated heat capacity is not taken to be zero. Therefore, the expressions for the enthalpies of formation of positive and negative ions reduce to:
ΔfH;(M+)298 K = ΔfH°(M)298 K + [IEa + C]
ΔfH;(M−)298 K = ΔfH°(M)298 K − [EA + C]
(where the quantity in brackets is the assumed enthalpy change of reaction at 298 K). What most often causes confusion for non-thermodynamicists is that the integrated heat capacity of the electron, C, becomes an increment in the value for the enthalpy of formation of the ion M+ or M−.
In contrast, the standard treatment of ion enthalpies of formation followed in almost the entire corpus of literature on ion physics/chemistry — the Ion Convention — is equivalent to assuming that the integrated heat capacity of the electron, C, is equal to the enthalpy of formation of the electron at temperature T, so that the equations reduce to:
ΔfH;(M+)298 K = ΔfH°(M)298 K − ΔfH;(e−)298 K + [IEa + C]
ΔfH;(M+)298K = ΔfH°(M)298 K + IEa
and
ΔfH;(M−)298 K = ΔfH°(M)298 K + ΔfH;(e−)298 K − [EA + C]
ΔfH;(M−)298 K = ΔfH°(M)298 K − EA
Thus, for the purposes of deriving enthalpies of formation of ions from ionization energy or electron affinity data, it does not matter what value is chosen for the integrated heat capacity of the electron, C, since it does not appear in the final expression for the enthalpy of formation. However, the expressions relating the enthalpies of reaction at finite temperatures to the zero degree quantities, IEa and EA, do explicitly include the integrated heat capacity of the electron:
ΔHI = IEa + C
ΔHEA = − EA − C
or, more correctly:
ΔHI = IEa + C + B − A
ΔHEA = − EA − C − A + D
As mentioned above, it is recommended that the value for the integrated heat capacity term for the electron derived using Fermi-Dirac statistics (3.145 kJ/mol, 0.0326 eV) be used.
Since in most thermodynamic literature or data compilations where the Electron Convention is used [6], values of enthalpies of formation of ions have been derived assuming that the heat capacity of the electron can be estimated to be that of an ideal gas following Boltzmann statistics, or 5/2 RT (6.197 kJ/mol at 298 K), the relationship between 298 K enthalpies of formation of ions in the Ion Convention (IC) and most published values for enthalpies of formation derived using the Electron Convention (EC) is:
ΔfH;(M+)298 K (IC) = ΔfH;(M+)298 K (EC) − 6.197 kJ/mol
ΔfH;(M−)298 K (IC) = ΔfH;(M−)298 K (EC) + 6.197 kJ/mol
In preparing the current edition of this data collection for distribution via the WebBook, the possibility was considered of using the Electron Convention. However, the decision was made to retain the use of the Ion Convention. The reasons for this decision were:
- The Ion Convention is to be recommended because of its simplicity. Introduction of an arbitrary (even if small) temperature dependence to ion enthalpies of formation invites confusion and error, especially among the large community of users/generators of such data who are accustomed to the Ion Convention.
- There is a great body of numeric data in the literature that was derived over the last 50 or more years using the Ion Convention. If the convention were changed, anyone using data from the older literature would have to make appropriate changes to these data before using them. Some error and confusion would inevitably result.
That is, it appears that considerable confusion could result from an attempt to change the presentation of data here. Therefore, in this database, the use of the "Ion Convention" is retained. It is recommended that in all literature dealing with thermochemistry of ions in the gas phase, clear signposts should be provided to indicate which convention is being used.
INTERPRETATION OF ION THERMOCHEMISTRY DATA
This section provides brief descriptions of some of the factors relevant to the interpretation and evaluation of ionization energy and appearance energy data. More detailed discussions of the ionization process are available in many books and reviews, notably in the Introduction to "Energetics of Gaseous Ions" [3]. Material presented here has as its focus those aspects of the subject which have a bearing on the evaluation of data on ionization energies, appearance energies, or ion/molecule equilibrium constants.
Ionization Thresholds
The Franck-Condon Principle
Ionization of a molecule by photoionization or by energetic electrons (sometimes called "electron impact") is governed by the Franck-Condon principle, which states that the most probable ionizing transition will be that in which the positions and momenta of the nuclei are unchanged. Thus, when the equilibrium geometries of an ion and its corresponding neutral species are closely similar, the energy dependence of the onset of ionization will be a sharp step function leading to the ion vibrational ground state. However, when the equilibrium geometry of the ion involves a significant change in one or more bond lengths/angles from that of the neutral species, the transition to the lowest vibrational level of the ion is no longer the most intense, and the maximum transition probability (the vertical ionization energy) will favor population of a higher vibrational level of the ion; if the geometry change is great, it is possible that the transition to the lowest vibrational level of the ion (i.e. the adiabatic ionization energy) will not even be observed. These situations are illustrated for hypothetical diatomic species in Fig. 1.
In evaluating ionization energy data, the shapes of photoelectron bands are useful indicators as to which of the situations pictured in Fig. l prevails for the particular molecule. A sharp onset indicates that the equilibrium geometries of ion and neutral are quite similar, and that photoionization or electron impact determinations of the ionization threshold are likely to be free of complications. When an ionization process proceeds according to the second situation pictured in the figure, the onset of the photoelectron band is observed approximately at the adiabatic ionization energy; adiabatic ionization energies derived from observation of the onsets of photoelectron bands are usually in excellent agreement with adiabatic ionization energies obtained from optical spectroscopy (analyses of Rydberg series) or from the most reliable threshold determinations.
When the equilibrium geometry of the ion is very different from that of the corresponding neutral molecule and the lowest vibrational level is not populated in ionization by photon absorption or electron ionization, it has been shown that values for the adiabatic ionization energies can be obtained by determining the equilibrium constant for charge transfer to another molecule of known ionization energy:
A+ + B ⇌ B+ + A
In such determinations, the ions are at thermal equilibrium with their surroundings, and one measures the thermochemical properties of the ions in their equilibrium geometries. The enthalpy change for this reaction, which is obtained from the equilibrium constant determination, is just the difference between the enthalpies of ionization, ΔHI, of species A and B. As derived in the discussion of thermochemistry of ions at finite temperatures, this difference is likely to be quite close to the difference in the adiabatic ionization energies:
ΔH = [ΔHI (B) − ΔHI (A)] ~ [IEa (B)-IEa (A)]
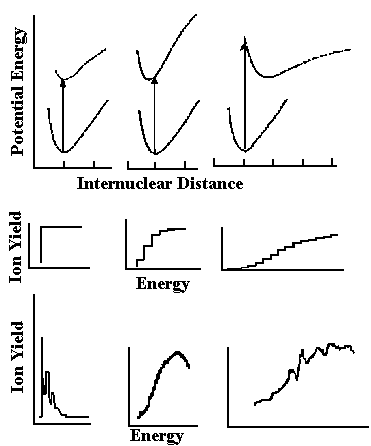
Fig. l. Potential energy curves for hypothetical diatomic molecule AB, and the corresponding positive ion, AB+ for the cases in which the equilibrium internuclear distance is (a) the same, (b) slightly different, or (c) greatly different. Below the potential energy curves are hypothetical probabilities for ionization as a function of energy for cases (a), (b), and (c), and, at bottom, shapes of observed photoelectron bands for the three corresponding cases.
Hot Bands
In addition to the problem of accurately detecting an ionization onset when there is a large change of molecular geometry in the ionization process, there is the problem of "hot bands". This term is applied to the observation of ionization at energies below the adiabatic ionization energy when there is a significant population of vibrationally excited molecules in the neutral species.
Appearance Energies
The appearance energy (sometimes called the appearance potential) is the minimum energy required to form a particular fragment ion from a precursor neutral molecule:
AB → A+ + B + e− ΔHrxn = AP
We have discussed elsewhere the derivation of the enthalpy of formation of the fragment ion, A+, from the appearance energy, and considerations having to do with derivations of enthalpies of formation at temperatures other than absolute zero. In those discussions, we specified that thermodynamic information can be obtained from appearance energy values only when there is no potential barrier in the reaction coordinate for the fragmentation reaction, and no significant "kinetic shift" associated with the determination (except for those experiments specifically designed to determine the kinetic shift). The meanings of these terms is given here.
Potential Barriers
It is well known that some ionic decomposition processes occur via pathways involving the surmounting of a barrier on the potential surface. This is especially true of rearrangements, in contrast to simple bond cleavage fragmentations. Surmounting such a barrier requires an activation energy greater than the enthalpy of reaction for the process. For this reason, the determination of thermochemical information from fragmentation threshold energies is always subject to the uncertainty that the enthalpy of reaction may not be the same as the observed onset energy. In some instances, the existence of a potential barrier for a fragmentation process may be deduced from experimental observations, such as fragment ion kinetic energies, or the shapes of metastable peaks.
Kinetic Shift
The "kinetic shift" is the term applied to experimentally-observed ionization onsets which are higher than the thermodynamic onset energy due to the fact that the apparatus samples the (fragmenting) ions at a certain time (usually around l0-5 s) after ionization has occurred, when ions undergoing a slow fragmentation process have not yet had time to dissociate. One approach for getting around this problem is an analysis based on the determination of the so-called rate-energy curve for a given fragmentation; in this approach, the rate constant of the dissociating ion is determined as a function of energy. The information is derived from an analysis of the data from a technique which is, moreover, capable of delivering very accurate thermochemical information for fragmentation processes, photoion-photoelectron coincidence spectroscopy (PIPECO). In addition, recent studies have approached this problem by sampling the fragmenting ions as a function of time using time-resolved dissociation techniques.
Rydberg Series
The interpretation of optical spectra to give ionization energies is a highly developed field with an extensive theoretical foundation. To summarize briefly, an atom with one excited electron in an orbital of high principal quantum number can, in first approximation be considered to be hydrogen-like. The excited electron is subjected to the central force field of the nucleus screened by the remaining core electrons. The energy levels of this system can be described by the Rydberg formula:
Tn = I − [2π2 Z2 e4 / ch3 n2 ] 7 [mM/(m + M)] = I − RZ2 / n2
where I is the ionization energy, Z is the net charge of the nucleus and core electrons, n the principal quantum number, e the electronic charge, m the electron mass, M the mass of the nucleus plus core electrons, c the velocity of light, h Planck's constant and R the Rydberg constant (for mass M). Although highly accurate ionization energies can be derived for atoms from Rydberg series, there are some possible complications such as the existence of terms and series which are perturbed by configuration interaction, or spin-orbit splitting of terms.
The identification of molecular Rydberg series is often not completely straightforward. Criteria that are used include the requirement that the transitions be strong and gradually decrease in intensity with increasing energy, and, evidently, that the series fit the Rydberg formula. Although some highly accurate molecular ionization energy data are derived from optical spectroscopic studies, especially for diatomic molecules, the evaluation of molecular Rydberg series is not always straightforward, and reported spectroscopic ionization energies of polyatomic species may disagree with values derived from ionization onset determinations or the onsets of photoelectron bands due to complications in the analysis of vibrational and rotational structure [14].
Ion/Molecule Equilibria
Most of the gas phase basicity / proton affinity and gas phase acidity data, as well as some of the electron affinity and ionization energy data presented in the WebBook collection are derived from determinations of the equilibrium constants for ion/molecule reactions:
A+ + B ⇌ C+ + D
A− + B ⇌ C− + D
Since the equilibrium constant
Keq = [C+ ][D] / [A+ ][B]
Keq = [C− ][D] / [A− ][B]
directly gives a value for the Gibbs free energy change associated with the reaction
- RTlnKeq = ΔG° = ΔH° − T ΔS°
relative thermochemical scales are generated through such measurements. In order to determine the enthalpy changes of these reactions, values for the entropy changes of reaction must be obtained, through measurements of the equilibrium constant as a function of temperature (Van't Hoff plot) or through statistical mechanical estimations.
In practice, many studies have been published in which measurements were made at a single temperature, with the (usually small) entropy change for the reaction estimated from statistical mechanical considerations. More recently, studies have appeared which include determinations of the equilibrium constant as a function of temperature (e.g. experimental determinations of the entropy changes), but in a recent evaluation [10] of gas phase basicity data, it has been found that Van 't Hoff plots determined in different laboratories usually show poor agreement, indicating possible experimental problems in these determinations.
This, in part may be due to a problem described [15] as pertaining to accurate determination of the reactive neutral pressure in pulsed high pressure mass spectrometers. The pressure in the source of such an instrument has often been in the transition regime between molecular and viscous flow. This can cause appreciable fractionation of neutral species in the bath gas, based on the diffusivity of the compound, which is related to the square root of the mass. Thus, both relative and absolute neutral pressures may have been poorly characterized in such experiments in the past. These could vary from instrument to instrument, based on design, and from experiment to experiment, based on varying operating conditions. Other possible contributing factors are clustering of neutral molecules to the ions at low temperatures, and pyrolysis of the ions at high temperatures.
Therefore, at the present time, it appears that the most reliable values for entropy changes associated with such ion-molecule equilibria can be obtained by judiciously examining experimentally-determined entropy changes in conjunction with ab initio calculations of those quantities.
Thermochemical scales derived from equilibrium constant determinations are, of course, scales of relative thermochemical values. Absolute values for thermochemical quantities can be assigned if a reliable value for the quantity in question (ionization energy, electron affinity, proton affinity, acidity, etc.) is available for one or more species in the scale.
The evaluation of thermochemical scales derived from equilibrium constant determinations presents special challenges, since the data for different molecules are all interrelated, so the scale must be evaluated as a whole, not molecule-by-molecule. That is, evaluation of such a thermochemical scale imposes the requirements of internal consistency in three parameters, Δ G (at different temperatures), Δ H and Δ S; furthermore, final absolute values assigned for properties of individual compounds must be consistent with what is known about enthalpies of formation of the relevant species, or with entropies of reaction that would be predicted from statistical mechanics and values (when available) of absolute entropies of the relevant species. In addition, trends in data for homologous series or compounds of a particular structural type must make sense.
The major uncertainty in data derived from equilibrium constant determinations, aside from the question of whether thermodynamic equilibrium is actually attained, is in knowing the temperature accurately. In the recent re-evaluation of the extensive scales of proton affinity and gas basicity, it was found that for the large body of data evaluated in the 1984 publication [8] the thermochemical scales (particularly the end of the scale representing high gas phase basicities) had been significantly constricted because operating temperatures of the instruments used in the experiments had been underestimated. For this reason, users will find that certain cited values for proton affinities and gas basicities given here are significantly different from those listed earlier [8, 9], although for all species for which sufficient information was available to do a complete evaluation, internal consistency is maintained.
Most ion/molecule equilibrium studies have been devoted to the derivation of extensive scales of relative proton affinities and gas phase acidities. The results were mainly derived from interlocking ladders of enthalpy changes for the proton transfer reactions:
AH+ + B ⇌ BH+ + A
AH + B− ⇌ A− + BH
Other published ion/molecule equilibrium studies provide data on charge transfer:
A+ + B ⇌ B+ + A
A− + B ⇌ B− + A
which yield scales of relative enthalpies of ionization or electron affinities at finite temperatures. A thermochemical ladder of relative ionization energies determined in this way [16] closely reproduces the equivalent scale of spectroscopic ionization energies, thus demonstrating the reliability of the approach for deriving information on relative ionization energies.
The most useful application of this approach for ionization energy data has proved to be the determination of ionization energies for species which undergo a large change of geometry upon ionization, and which therefore exhibit very gradual onsets of ionization as a function of energy. For example, the only reliable information about the adiabatic ionization energies of n-alkanes and of alkyl hydrazines comes from thermochemical ladders established through equilibrium constant determinations. Since the electron transfer occurs in a long-lived collision complex which endures for many vibrational periods, the ionic configuration corresponding to the equilibrium geometry of the ion (the geometry corresponding to the adiabatic transition) is accessed.
Other equilibrium studies have been concerned with hydride or halide transfer reactions:
R1+ + R2X ⇌ R2+ + R1 X
(where X is H, F, Cl, Br, or I). Studies of hydride transfer and halide transfer equilibria have led to quantitative information about the relative enthalpies of formation of alkyl carbocations. These data were used to supplement information from appearance potential determinations in evaluating enthalpies of formation of alkyl carbocations.
See the discussion of determinations of ion/molecule equilibrium constants for more experimental details.
Thermochemistry of Ions in the Gas Phase at Finite Temperatures
The auxiliary thermochemical information required for citation of ion enthalpies of formation — enthalpies of formation of relevant neutral species - is available mostly for species at 298 K. These thermochemical data are correct for use in treatment of data for processes occurring at temperatures other than 0 K (ion/molecule equilibria, for example). However, strictly speaking, ionization energies, appearance energies, and electron affinities are quantities which correspond to processes occurring at 0 K. To correctly derive enthalpies of formation of ions from these kinds of data requires explicit treatment of the differences in thermochemical values at 0 K and at higher temperatures. This section discusses the principles involved in such a correct treatment, describes the simplifications which are often made in the literature, and specifies how data are treated here.
Molecular Ions
The enthalpy of formation of molecular ion M+ or M− at temperature T can be defined in terms of the enthalpy of formation of the corresponding neutral species, M, at temperature T, and the enthalpy change of the ionization process at that temperature, ΔHI or ΔHEA for positive or negative ions, respectively.
ΔfH;(M+)T = ΔfH°(M)T − ΔfH;(e−)T + ΔHI
ΔfH;(M−)T = ΔfH°(M)T + ΔfH;(e−)T + ΔHEA
In applying this equation, the values for ΔHI or ΔHEA are usually taken to be exactly equal to the adiabatic ionization energy or negative of the electron affinity. Actually, however, the ionization energy and electron affinity are the equivalents of the spectroscopic transitions between the lowest rotational and vibrational levels of the ground state of the molecule and the lowest rotational and vibrational levels of the electronic ground state of the ion; that is, they are equal to the difference between the enthalpies of formation of the molecule and the corresponding ion at absolute zero. As derived in the discussion on Thermochemical Conventions, the enthalpy changes of reaction at temperature T are related to the adiabatic ionization energy and electron affinity by the expressions:
ΔHI = IEa + (C + B − A)
ΔHEA = − EA − (C + A − D)
where A, B, D, and C represent, respectively, the integrated heat capacities (energies required to raise the species from 0 K to temperature T) of the neutral molecule (A), positive (B) or negative ion (D), and electron (C). Thus the expressions for the enthalpies of formation of the ions at temperature T become:
ΔfH;(M+)T = ΔfH°(M)T − ΔfH;(e−)T + IEa + C + B − A
ΔfH;(M−)T = ΔfH°(M)T + ΔfH;(e−)T − EA − C − A + D
Using the Ion Convention where ΔfH;(e−)T is taken to be equal to C, the full expressions for deriving a value for an enthalpy of formation of an ion at temperature T using the adiabatic ionization energy or electron affinity and values for ΔfH;(M+)T or ΔfH;(M−)T reduce to:
ΔfH;(M+)T = ΔfH°(M)T + IEa + B − A
ΔfH;(M−)T = ΔfH°(M)T − EA − A + D
Thus, for a correct treatment of the enthalpy of formation of an ion at any temperature other than absolute zero, the integrated heat capacities of the ion, B or D, and its precursor neutral molecule, A, (or more specifically the differences between those heat capacities) must be taken into account. However, it is a common practice to derive "298 K heats of formation" of positive ions by simply adding the 0 K value for the ionization energy to the 298 K enthalpy of formation of the molecule. Therefore, we must examine to what extent the simplifying assumption that A = B = D is valid.
A published analysis of the differences between integrated heat capacities of M and M+ for various molecules demonstrated [16] that (a) there will be no discernible differences between the translational and rotational heat capacities of M and M+, (b) that differences arising from a splitting of degenerate energy levels in multiplet ground states of M or M+ will never be larger than 0.009 eV (0.9 kJ/mol) at temperatures in the 300–400 K range, and (c) when the frequency of a particular vibration changes upon ionization, a difference between the integrated heat capacities of M and M+ will result. However, even this contribution will usually be sufficiently small that a significant error will not be introduced if it is ignored. For example, the lowest ionization energy of ethylene corresponds to removal of an electron from the C-C pi bond, which leads to a lowering of the frequency of the symmetric C-C stretch from 1623 to 1230 cm-1 and a reduction in the frequency of the twisting around the C–C bond from 1027 to 430 cm-1. Although these differences in vibrational frequencies are significant, the predicted effect on the 298 K enthalpy of ionization is to raise it above the value for the adiabatic ionization potential by only 0.0069 eV (0.7 kJ/mol), i.e. only the most accurate experimental measurements could detect an increment of this size. Although this analysis was concerned with heat capacities of positive ions, the same reasoning — and the conclusions — should apply equally well to negative ions, that is, to changes in the electron affinity with temperature.
Therefore we conclude that for most species, the simplifying assumption that the adiabatic ionization energy or electron affinity and the corresponding enthalpies of ionization at an elevated temperature are approximately the same:
IEa ~ ΔHI
-EA ~ ΔHEA
will introduce errors in enthalpies of formation derived for molecular ions at 298 K of the magnitude of 2 kJ/mol or less. At higher temperatures, it is possible that larger errors would result from this simplification.
In this database, most of the cited values for enthalpies of formation of molecular ions correspond to 298 K, and were obtained by simply adding the value for the adiabatic ionization energy to the 298 K enthalpy of formation of the neutral species; that is, the assumption discussed above was usually made. A rigorous treatment would require calculating exact values for integrated heat capacities and from complete sets of vibrational frequencies for the molecule and the ion. Vibrational frequencies for most of the ions are not available, and the correction would simply cancel out if one made the often-used assumption that the vibrational frequencies of the ion and its neutral counterpart are the same. Whenever the original authors of a paper carried out such a complete analysis (a routine procedure only for photoelectron-photoion coincidence studies), the results of that analysis are included here, and both 0 K and 298 K values for the ion enthalpy of formation are given; only in these cases is the temperature specified.
Fragment Ions
Arguments analogous to those given for molecular ion data can be applied to the use of appearance energies for the derivation of enthalpies of formation of fragment ions, A+, at temperature T. If there are no complicating factors (such as potential barriers or a kinetic shift), the appearance energy, AP, corresponds to the enthalpy change for the fragmentation reaction, and can be used to derive a value for the enthalpy of formation of the fragment ion, A+.
ΔfH;(A+) = ΔfH°(AB) − ΔfH°(B) − ΔfH;(e−) + AP
Correctly, a 0 K enthalpy of formation of A+ must be obtained using 0 K enthalpies of formation of AB and B in the calculation, and this enthalpy of formation can then be corrected to some other temperature, T, taking into account the vibrational frequencies of the ion and appropriate thermodynamic functions of the elements.
For the most common experimental techniques (energy selected electron impact, photoionization techniques, etc.) used to measure the appearance energy of a fragment positive ion starting from a molecule or radical at temperature, T, the major problem is to identify the internal energies of the reaction products. This matter has been discussed at length by Traeger and McLaughlin [17]. At onset the products of the unimolecular decomposition will be formed with zero translational energy with respect to the center of mass (provided that the fragmentation does not involve a reverse energy barrier) and a center of mass translational energy the same as that of the precursor molecule. The products thus are at a translational quasi-temperature, T *. In principle, if the observational time scale of the experiment and the sensitivity of the ion detector are great enough, then the observed appearance energy approaches that for products having 0 K internal energy (i.e., all internal energy modes have contributed to reaching the transition state). Traeger and McLaughlin showed that for the molecule RX dissociating to give (R+ + X):
AP T (exp) = ΔfH; [R + + X + e] T − ΔfH° [RX] T − 5/2RT − ∫ Cp [R + + X + e] d T
In effect, this equation corrects the observed threshold energy for the fragmentation process to an effective 0 K value by adding the thermal rotational and vibrational energy contained in RX to the onset energy.
Most enthalpies of formation of fragment ions are derived making the simplifying assumption that the last two terms of this equation will cancel one another. That is, values for enthalpies of formation of fragment ions at 298 K derived from appearance potential data are more often obtained by simply using an observed onset energy and 298 K enthalpies of formation of relevant neutral species.
ΔfH;(A+) = ΔfH°(AB) − ΔfH°(B) − ΔfH;(e−) + AP
The user should be cautioned that the 298 K value assigned to a enthalpy of formation of a fragment ion may differ by as much as 12-18 kJ/mol, depending on which of these treatments has been used. For example, Brand and Baer [18], and Lossing [19] determined the appearance energies for formation of C4H7+ ions in C5H10 isomers. Although the appearance energies reported in the two studies were almost identical, the 298 K values for enthalpies of formation of the C4H7+ ions derived by Brand and Baer, using a complete treatment of the temperature dependence of the enthalpy of formation, are higher than the values derived by Lossing by 18 kJ/mol.
As mentioned elsewhere, this database displays evaluated values for enthalpies of formation for only a very few much-studied fragment ions at the present time; as evaluations are made, additional values will be added. However, in many cases, sufficient information is given in the WebBook databases to allow users to derive such values as needed.
EXPERIMENTAL TECHNIQUES
In this section, we summarize briefly the different types of experiments from which the data presented here originate, and where possible indicate the strengths and/or limitations of the different techniques, and how these influence the evaluator in arriving at a recommended value for a particular property.
In the database, techniques are identified by acronyms, shown in square brackets in the discussion, or in some cases, after the appropriate headings. A problem in defining an experimental technique for the purposes of assigning acronyms in the database is that acronyms used in the literature have evolved over the years as experimental techniques evolve, so that for a collection like this one with experimental results originating over a 70-year time period, a problem of internal consistency arises. Because of this problem, we have elected to use fewer, more broadly-defined acronyms, rather than attempt to maintain a detailed breakdown of experimental techniques, which may differ only in subtle details, in the assignment of acronyms. Even using broadly-defined acronyms, there are certain studies for which it is difficult to pigeonhole the experimental technique; for example, the borderline between "photoionization" and "laser spectroscopy" is sometimes not easily defined, and the distinction is sometimes made more on the basis of the focus of the study than on the actual details of the experiments.
For at least two specialized techniques that would logically fit under our broad umbrella-acronyms, we have assigned individualized acronyms, because the data provided by these techniques — time-resolved photodissociation (TRPI) which is actually a photoionization (PI) technique, and photoion-photoelectron coincidence (PIPECO) − which could also be considered either photoionization (PI) or a threshold electron detection (TE) technique − are uniquely informative. In the case of time-resolved photodissociation results, measured appearance energies often differ significantly from those determined using other techniques since slow fragmentation processes are detected.
Optical Spectroscopy [S]
The identification of a Rydberg series in an atomic or molecular spectrum leads to a value for the ionization energy. In cases where the analysis of the spectrum is straightforward, the spectroscopic ionization energy values are highly accurate. The determination of atomic ionization energies through optical spectroscopy is a highly developed field which has been extensively reviewed. A large fraction of atomic ionization energies listed here are from expert evaluations of atomic spectra. In the evaluation of ionization energies of atoms and diatomic molecules, spectroscopic ionization energies have been chosen as "selected values" where they are available. For polyatomic species, a value derived from an analysis of the optical spectrum has been given great weight, unless several determinations from other highly reliable techniques are in conflict with the spectroscopic value.
Threshold Experiments
The most widely-used technique for determination of ionization and appearance energies involves a direct determination of the minimum energy required to form a parent or fragment ion from a neutral species, or to detach an electron from a negative ion. In these approaches, ionization may be effected by photoionization, by interaction with energetic electrons ("electron ionization" or, in older literature, "electron impact"), or by interaction with excited atoms (Penning ionization) or other chemi-ionization reactions; the measurement involves a determination of the minimum energy ("threshold") required to form an ion. The resulting ions or the ejected electrons, or both, are detected using various mass spectrometric techniques.
Electron Ionization Techniques[EI],[EIAE]
Over the years, the most widely-used techniques for the determination of ionization and appearance energies have involved the use of mass spectrometers in which ionization is effected by an electron beam. The energy of the beam is varied, and the abundance of the resulting ion(s) is monitored; the "onset" of the ion on the energy scale must be detected. A problem with this approach that had to be dealt with before accurate data could be obtained was that standard electron beams had a large energy spread, so the nominal energy expected from the applied electrode potentials was not a good indication of the actual energy of electrons in the beam. However, by the 1960s, techniques were developed [20] to narrow the energy range of the electron beams through the use of so-called "electron monochromators", in which the energy of the electron beam is narrowly defined by passing the beam through electron energy selectors of various designs. Other laboratories have utilized a so-called fast-beam apparatus to determine accurate ionization cross sections as a function of electron energy. Modern results obtained using electron beams with well-defined energies are in excellent agreement with analogous results derived from determinations of photoionization thresholds.
Early on, there were other, somewhat less rigorous, approaches to solving this problem of energy spread in electron beams, leading to what Rosenstock et al [3] call "quasi-monoenergetic" beams (the "Retarding Potential Difference" [21] and "Energy Distribution Difference" [22] methods).
Some electron ionization data reported here were never intended to be accurate ionization energy/appearance energy determinations, but were carried out simply as diagnostic measurements, to unravel chemical processes occurring in particular systems (such as in the vapor over a Knudsen cell).
In the WebBook database, all of these techniques are simply designated by the acronym "EI" or for negative ions "EIAE". Measurements made using the more careful approaches can be distinguished from the non-quantitative measurements by the cited error limits or (where there are no error limits given) by the number of significant figures displayed. Most of the quantitative measurements made during the past 20 years have been made with well-defined electron energies.
Photoionization Techniques
Photoionization Mass Spectrometry [PI],[PD],[LPD]
In the mid-1960s, the problem of exactly defining the energy of the ionizing agent was approached in some laboratories by replacing the electron beam by photons, whose energy could be well defined. In classical photoionization mass spectrometry, monochromators were used with standard light sources, and the ion abundance was determined as a function of photon energy. That is, the approach to determining a threshold energy was exactly analogous to that used in electron ionization experiments. In such experiments, as with the electron ionization techniques, one must be able to detect the onset of ionization. According to the Franck-Condon Principle, if the configuration of the ion is different from that of the precursor neutral molecule, the onset of ionization as a function of energy will be gradual, and the exact onset may be difficult to pinpoint accurately.
Modern photoionization experiments often utilize laser or synchrotron light sources, and may have other distinctive features designed to provide more detailed information. For example, studies are published examining pressure- or time-dependencies of ionic photodissociation processes. The latter are assigned a separate acronym here (TRPI), rather than being categorized with other "photoionization" experiments, since observed onsets may differ markedly from onsets measured on the conventional time-scale, and it is useful to be able to recognize (through the acronym) why this is so.
The electron affinity of a species can be determined by finding the threshold for electron photodetachment, via irradiation of a trapped negative ion by variable frequency light. This is denoted by the acronyms [PD] for photodetachment, using a continuous frequency light source and a monochromator, or [LPD] for use of a variable frequency laser as the light source.
Time-Resolved Photodissociation [TRPI]
Two complementary photoionization methods are often used for studying dissociation processes where a "kinetic shift" exists (that is, where experimentally-observed ionization onsets are higher than the thermodynamic onset energy due to the fact that the apparatus samples the fragmenting ions at a certain time when ions undergoing a slow fragmentation process have not yet had time to dissociate). These approaches, both of which examine the dissociation process as a function of time, are time-resolved photodissociation (the acronym TRPD is used in the literature) and time-resolved photoionization mass spectrometry (the acronym TPIMS is used in the literature). In time-resolved photodissociation experiments, parent ions are formed by electron impact, thermalized, and photoexcited by a monochromatic pulsed laser; the dissociation is followed as a function of time in an ion cyclotron resonance spectrometer (ICR). In time-resolved photoionization mass spectrometry, ions are formed by a pulsed VUV light source in an ion trap, and are ejected after a given delay time into a quadrupole mass filter. While both approaches give time-resolved information about dissociation processes, they are complementary rather than effectively the same since in the former technique, all parent ions are excited to the same energy, while in the latter, parent ions are excited to a range of internal energies extending from zero up to the maximum available energy.
Laser Spectroscopy [LS]
(Multi-Photon Ionization and Resonance Ionization Mass Spectrometry)
In recent years, spectroscopic studies using laser techniques have provided highly accurate ionization energy values. For example, ionization energies for molecules have been determined using multiphoton ionization or resonance-enhanced multiphoton ionization (REMPI) of vibrationally-cooled species in a molecular beam. In these studies, the cooled beam of molecules is raised to a specific excited state by irradiation with a tunable laser; while this excitation energy is held constant, a second independently tunable laser is used to ionize the beam of excited molecules, with the photon energy being tuned through the ionization onset. The excitation laser is then tuned to a different transition, and the ionization scan is repeated. In this way, the entire Franck-Condon accessible region of the intermediate electronic state is mapped out, insuring that the molecular geometry corresponding to the adiabatic ionization energy is accessed. Since every intermediate vibronic state leads to an independent value of the ionization threshold, the experiment contains an internal consistency check.
For atoms, the related technique known as Resonance Ionization Mass Spectrometry (RIMS) is sometimes employed to determine accurate ionization energies.
Photoelectron Spectroscopy [PE], [LPES]
It is also possible to determine the energy change associated with an ionization process by effecting ionization with a photon of well-defined energy and measuring the energy of the ejected electrons:
M + hv → M+ + e−
where
KE(e−) = hv − I − E*(vib,rot)
(where E*(vib,rot) is the internal energy of M+ and I is the binding energy of the electron).
The most widely-used technique of this type is conventional photoelectron spectroscopy in which the photon sources are usually the helium or neon resonance lines (21.218 eV and 40.813 eV or 16.848 eV and 16.671 eV, respectively) or other intense monochromatic sources. In such an experiment, the ejected electrons will have differing energies depending on the distribution of energy levels in the M+ ions formed; a map of the abundances of the ejected electrons as a function of energy is called the photoelectron spectrum. The shapes of the photoelectron bands will reflect not only the energy differences in the different states of M+, but also show the M → M+ transition probabilities as governed by the Franck-Condon principle. In cases where the equilibrium geometry of the ion and the corresponding neutral are the same or are similar, it is found that the observed onset of the first photoelectron band is usually a reliable indicator of the adiabatic ionization energy. This situation is easily recognized by the sharp onset of the photoelectron band.
Most photoelectron spectroscopy studies are carried out with the goal of elucidating the spectroscopy of the system through determinations of vertical ionization energies leading to the ground state and excited state ions, and therefore, little attention is usually given to determinations of adiabatic ionization energy values. In many instances, figures showing the photoelectron spectra are displayed, and onsets of photoelectron curves which correspond approximately to the adiabatic ionization energies can be estimated from the figure. Such values are reported here, but are surrounded by parentheses to indicate that these are approximate values not selected by the original authors.
For negative ions, if a beam of ions is irradiated by a laser beam with photons in excess of the energy required to detach the electron, then analysis of the translational energy of the detached electrons allows for determination of the electron affinity. This technique [LPES] can provide electron affinities precise to micro-electron volts. The method often provides information on the vibrational states of the neutral and ionic species as well. However, the assignment of the (0-0) threshold can be complicated by these states. The precision is commonly better than 0.2 kJ/mol, and can be much better.
Threshold Electron Detection [TE], [ZEKE]
Highly accurate determinations of ionization energies come from a family of techniques in which laser ionization (e.g. REMPI) or pulsed field (PFI) techniques are combined with the detection of energy-selected electrons. In so-called "threshold photoelectron spectroscopy" or "zero-kinetic energy spectroscopy" (the acronym "ZEKE" is often used in the literature) ions with a well-defined energy are formed, and only those electrons which correspond to essentially zero energy of ejection are detected. Such experiments yield vibrationally/rotationally resolved spectra of the ions. In some cases, mass analysis of the positive ion is included (mass-ZEKE). The acronym [ZEKE] is used in the negative ion data base here.
Photoion-Photoelectron Coincidence Spectroscopy [PIPECO]
For the purposes of studying the thermochemistry of ionic fragmentation processes, a powerful variation of the Threshold Electron Detection approach is used which involves the simultaneous detection of a (single) zero-kinetic energy electron and the corresponding (single) positive ion. In the technique known as photoion-photoelectron coincidence (or sometimes, photoelectron-photoion coincidence — PEPICO), ejected electrons which originated with "zero" kinetic energy are matched with their corresponding positive ions. At energies where parent ions, M+, are undergoing dissociation to form one or more fragment ions, one obtains the relative probabilities for the formation of the daughter ions from parent ions of exactly known energy (i.e. the breakdown curve). The ions can be detected at differing times after the ionization event for the determination of the time dependence of the dissociation process. The complete interpretation of such data requires a modeling of the dissociation using statistical theories of unimolecular decomposition (i.e. quasi-equilibrium/RRKM theory), but the thermochemistry and detailed mechanism of an ionic fragmentation process can be mapped out very accurately. As pointed out by Dannacher [23], in spite of its great strengths, this technique has not been widely utilized, possibly because of the intricate instrumentation required, the complexity of the data analysis, and the fact that each determination requires the investment of a great amount of time on the part of the experimentalist.
Chemi-Ionization [CI]
In the older literature, Penning Ionization — ionization by collisions with a beam of metastable neutral rare gas atoms with known excitation energy or energies — was often used as an ionization mechanism.
X* + M → M+ + X + e−
In these experiments, metastable atoms employed include mixtures of He(23S) (19.818 eV) and He(21S) (20.614 eV), Ne(3P2) (16.619 eV) and Ne(3P0) (16.715 eV), or Ar(3P2) (11.548 eV) and Ar(3P0) (11.723 eV). Because of the presence of two metastable states in a given atom beam, the Penning electron spectrum consists of a shifted superposition of two spectra, each formed by one of the species. The energy of the ejected electrons was analyzed.
Some recent studies have examined systems where other chemi-ionization reactions such as:
M + X2 → MX+ + X−
occur, and have obtained thermochemical data from experiments in which the collision energy is varied, and positive and negative product ions are counted and analyzed mass spectrometrically.
Charge Exchange Mass Spectrometry [CEMS]
In this technique, ionization is effected through charge exchange:
X+ + M → M+ + X
and a determination of the absolute cross section for production of M+. A series of charge donors, X+, of varying recombination energies (that is, where the ionization energy of X varies) are used. The plot of cross section as a function of recombination energy provides values for ionization and appearance energies. Because a continuous energy scale is not available with this technique, error limits are usually of the order of ±0.1 eV.
Neutral Beam Ionization or Appearance Energy [NBIE],[NBAE]
Collision of a neutral species with an energetic particle of low ionization potential, such as an alkali atom, can result in electron transfer, giving an alkali cation and an anion. The electron affinity of the neutral species is equal to the translational energy of the alkali atom less its ionization energy. Determinations of electron affinities by this method have the advantage that one obtains values for the true electron affinity: electron attachment to a neutral species, rather than detachment from an anion. Certain anions can be produced by this technique which are not accessible via electron impact due to low energy exit channels, e.g. CCl4 Due to the limited energy resolution of the neutral alkali beam, the precision of this technique is not high, typically 20 kJ/mol. The onset energies of fragment ions can also provide useful thermochemical information, if the thermochemistry of the coproduced neutral species is known. Normally this technique results in a determination of the adiabatic electron affinity, but for a sufficiently fast beam of neutral species, the onset corresponds to the vertical attachment energy of the electron, which, in contrast to detachment methods, is smaller than the adiabatic value.
Electric Field Detachment [EFD]
Negative ions with electron affinities of a few tenths of an electron volt or less can undergo detachment of the electron in a strong electric field. Based on the strength of the field, the dipole moment of the neutral species produced, and the rate of loss of the electron, the electron affinity can be derived. This often is used to access information on negative ions where the electron is in a non-valence state, such as dipole-bound anions.
Thermochemical Information from Studies of Ion/Molecule Reactions
Ion/Molecule Equilibrium Constant Determinations [EQ], [IMRE], [TDEq], [TDAs]
An ion/molecule equilibrium:
A+ + B ⇌ C+ + D
A− + B ⇌ C− + D
is established in a high pressure mass spectrometer, flow tube, or ion cyclotron resonance spectrometer, and the equilibrium constant is determined by observing the relative abundances of the two ions, A+/- and C+/- after a large number of collisions:
Keq = [C+/- ] [D] / [A+/- ] [B]
(where A+/- and C+/- are generic representations of positive/negative ions). The neutral reactants, B and D, are present in great abundance compared to the ionic reactants, and therefore, the ratio [D]/[B] does not change as equilibrium is established. A single measurement leads to a value for the Gibbs energy change of reaction at the temperature of the measurement, while a series of measurements at different temperatures permits an experimental evaluation of the entropy and enthalpy changes associated with the reaction:
- RTln Keq = ΔG° = ΔH° −TΔS°
The main uncertainty associated with this technique, aside from the necessity of ensuring that the system is at a true thermodynamic equilibrium, is that the temperature of the reacting system must be accurately known. Although the reproduction of relative spectroscopic ionization energies through equilibrium measurements carried out in widely different pressure regimes demonstrates that this is not a serious problem, it is true that the initially-reported networks of gas phase basicities led to thermochemical ladders that were constricted by as much as 15% because of problems with temperature determinations [10]. In addition, since the measurement leads only to relative thermochemical data, the resulting thermochemical ladders must be related to reliable comparison standards if absolute values are desired.
In the negative ion database, the acronym [IMRE] is used to indicate an equilibrium measurement made at a single temperature, while [TDEq] denotes those at multiple temperatures. [TDAs], for temperature-dependent association, is used in specific case of association equilibria such as:
A− + B ⇌ AB−
The positive ion database uses the identifier [EQ] for all such results.
Determinations of Reaction Endothermicity [END], [Endo]
Some thermochemical data reported here comes from studies where the enthalpy change of an endothermic ion/molecule reaction is determined. Information about the thermochemistry of a particular ion is obtained when relevant thermochemical data for other species participating in the reaction is available. In a very few instances, such information comes from straightforward kinetic treatments (Arrhenius plots) of the temperature dependencies of the rate constants of endothermic ion/molecule reactions. However, more commonly a so-called guided-beam apparatus is utilized for such determinations. Ions are generated in a flow tube, extracted and mass analyzed, then focused at the desired kinetic energy into a static cell containing the neutral reactant. The kinetic energy of the reactant ions is varied, and reaction onsets are determined as a function of translational energy.
Ion/Molecule Bracketing Experiments [IMB], [IMRB]
There are some ion/molecule systems for which an equilibrium can not be established in an ion source, either because one of the relevant neutral species is unstable (e.g. a radical or unstable molecule) or because of competing reactions in the system. In such cases, it is sometimes possible to obtain an experimental estimate of the enthalpy change of a particular reaction (charge transfer, proton transfer, hydride transfer, etc.) by use of a technique known as "bracketing" in which the ion of interest is reacted with a series of molecules chosen for variations in the relevant thermochemical parameter (ionization energy, electron affinity, gas phase basicity, acidity). The occurrence, and sometimes the rate constant, of reaction is monitored as a function of the parameter of interest. The approach is based on the assumption that an endothermic reaction will not be observed, or will occur only at a low efficiency. Thus, the approximate onset energy is usually assumed to lie on the energy scale at a point where the rate of reaction becomes very slow. One problem with this approach is that the reaction of interest may be exothermic (even highly exothermic) but will not be observed if another, more favorable reaction channel is available to the reacting pair.
In the positive ion database, only a few gas phase basicity values are derived from such measurements, but the information from these experiments is useful in evaluating conflicting experimental results from other techniques. More widespread use of this technique is made in the negative ion database.
The Kinetic Method [KIN], [BRAN], [CIDC]
If certain criteria of similarity in structure occur, the ratio of ions from the competitive fragmentation of an dimeric ion has been shown to reflect the thermochemical stability of the product ions. The energy for activation of the precursor ion can be from collision-induced dissociation, chemical activation, or the ion may be formed is a metastable state. Proton-bound species such as MeNH3+..H2NEt or MeOH..−OEt are examples of the structures applicable to this method. A calibration line based on the ion ratios of several species with energetics known from other methods, such as ion/molecule equilibria, is necessary to assign quantitative values in this experiment. Values denoted as [BRAN] for negative ions are from chemically-activated species; those labeled [BRAN] (positive ions) or [CIDC] (negative ions) are from metastable or collisionally-activated species.
The Electron Capture Detector [ECD]
An electron capture detector, using a beta source, can be modified to operate in a time resolved mode. By modeling the response of the ECD as a function of both time and temperature, the rate constants for both attachment and detachment of electrons can be determined, and thus the equilibrium constant for binding the electron can be obtained, leading to a value for the electron affinity. The method appears limited to EAs of 1 eV or so and less.
The Electron Swarm [ES]
The rate constant for electron attachment to a neutral can be measured as a function of mean electron energy in a drift tube. When combined with detachment rate constants from a beam technique, the electron affinity can be estimated. This gives a lower limit, which is often up to 0.5 eV too low.
Other Experimental Techniques
Essentially all of data included in this collection have been derived from results obtained using the experimental approaches listed above. Data are also included which result from the use of several additional techniques that are not easily placed in the broad categories as organized here.
Charge Inversion Energy Loss Spectrometry [CIEL]
Positive ions are generated by an electron beam and accelerated to translational energies well above thermal (4-6 eV), mass-selected and transmitted into a collision-gas cell, where they undergo collisions with an appropriate target gas. Electron capture reactions with the target molecules occur:
M+ + T → M + T+
M + T → M− + T+
Any stable anions that are formed are translational-energy analyzed prior to detection. As only forward-scattered anions are detected, the translational energy losses associated with these species are essentially equal to the endoergicities of their formative process.
Collisionally-Activated Dissociation [CAD], [CIDT]
Ions are generated, mass selected and accelerated into a chamber where they collide energetically with target molecules, and undergo collisionally-activated dissociation processes. The onset energies for particular dissociations are determined. Most of the body of data originating from this type of experiment has not been incorporated into the positive ion database, in that the quantity which is determined is the energy required to form a fragment ion from a precursor ion (rather than from a neutral species), and therefore, to match conventions used for presenting data in this database, the collisional onsets must be normalized by adding them to the appropriate ionization energy. In the future, this body of data will be re-examined, and, if there is sufficient demand, will be incorporated into an auxiliary database. There is extensive use of this method in the negative ion database, where the acronym [CIDT] is used.
Delayed Thermionic Ionization [DTI]
The one study [24] using this technique whose results are included in the database was an investigation of fullerenes. Ions and neutral molecules in the gas phase were generated by laser desorption from stainless steel target rods coated with C60 or C60 /C70 films. Delayed ionization by thermionic emission during the flight time between source and time-of-flight acceleration optics was detected by withdrawing ions perpendicular to the cluster beam. Thermionization rates were determined from these measurements, and ionization energies were derived from the results using statistical mechanical formulations. For details, users are referred to the original reference [24].
Charge Transfer Spectra [CTS]
The charge transfer spectrum method is a semi-empirical method often used, especially in the past, to estimate ionization energies of large molecules such as polycyclic aromatics and certain biochemical compounds. It is based on a semi-empirical theory developed by Mulliken [25] to explain the absorption bands of electron donor-electron acceptor complexes in solution. These bands arise from a transition from the ground state of the molecular complex to an excited state in which an electron is largely transferred from the donor to the acceptor molecule. The bands are not characteristic of the isolated donor or acceptor molecules. Hastings et al [26] derived from the Mulliken theory a simple algebraic relation between the frequency of the maximum of the charge transfer band and the ionization energy of the donor, and correlated it with experimental information. A limited comparison of ionization energies derived by this method and more accurate methods indicates that the estimates are usually within a few tenths of an electron volt of the more correct value.
Auger Electron Spectroscopy [AUG]
The technique of Auger Electron Spectroscopy is similar in principle to photoelectron spectroscopy. It is based on an analysis of the energies of ejected electrons. However, in this case the electron is ejected via an Auger cascade following prior inner shell ionization. The inner shell ionization is brought about by a high energy electron beam or a discrete X-ray source.
Surface ionization [SI]
The surface ionization method has been applied to the determination of first ionization energies of some metal atoms. The method is based on the assumption that the atoms in a beam, after impinging on a hot metal surface, will come to thermodynamic equilibrium, producing a surface concentration of atoms and ions whose composition can be described by the Saha-Langmuir equation:
N+ / N0 = g+ / g0 exp[e(φ − I)/kT
where N+ /N0 is the fraction of the atoms which are ionized, g+ and g0 are the statistical weights of the ions and atoms, e is the electronic charge, φ the work function of the metal, I the ionization energy, k the Boltzmann constant, and T the absolute temperature. The temperature dependence of the positive ion current gives the ionization energy if the work function is known. Complications include the effect of surface coverage or impurities on the work function, definition of the work function for a polycrystalline surface exhibiting a variety of crystal planes with different work functions, and an occasional lack of reproducibility of experimental results. Where comparisons can be made with more reliable methods, determinations of relative ionization energies using this approach reproduce other results within several tenths of an electron volt.
For negative ions, a common version of this experiment, the Magnetron technique, lacks mass analysis, and therefore many of the values for thermochemical parameters resulting from this method correspond to anions of uncertain identity. Precision is claimed to be several tenths of a volt (>20 kJ/mol), but appears to be worse in many cases, based on current data.
Liquid-Phase Electrochemical Determinations [LE]
Several studies have been published in which gas phase ionization energies are determined from measurements of photocurrent thresholds of compounds dissolved in nonpolar solvents. This approach was used primarily for obtaining ionization energy data for non-volatile organic compounds. The Born equation is employed to obtain the solvation energies for the cations.
Electron Auto-Detachment Rate [KINT]
For negative ions with electron affinities of less than about 0.5 eV, the rate for auto-detachment of the electron as a function of temperature can be measured. The activation energy obtained from this represents the electron affinity.
Electron Transmission Spectroscopy [ETS]
In this technique, the scattering of a monoenergetic electron beam impacting on a gas at less than the ionization threshold is determined. The presence of resonances in the spectrum implies electron capture to produce a temporary state, followed by autodetachment. This is the principal technique for measurement of negative electron affinities, i.e. cases where the anion is less stable that the neutral species. Occasionally, a series of resonances can be extrapolated to below zero electron energy to give an estimate of a positive electron affinity.
Kinetic Mobility of Ions [Mobl]
If the mobility of an ion in a gas can be measured in response to a weak electric field, the potential well depth, corresponding to the enthalpy for the ion associating with the neutral gas can be determined.
Laser Optogalvanic Spectroscopy [LOG]
The gas of interest is subjected to an electrical discharge, and the discharge region is probed by a laser. The 'LOG' spectrum is recorded by scanning the wavelength of the laser, and monitoring laser-induced changes in the discharge impedance. The spectrum produced will be similar to the laser absorption spectrum but relative intensities of spectral features may be very different. The method is particularly suitable for detecting unstable (radical) species.
Zero-pressure Thermal-radiation Induced Dissociation [ZTID]
In an Paul (ICR) trap, infrared radiation from the walls is absorbed by trapped ions. If a weak bond exists in the ion, the ion can dissociate to a new ion and neutral species, as its internal energy increases with time. Modeling of the rate constant for this process can yield a bond strength.
Scattering of Atoms from Ions [Scat]
If a beam of ions is passed through a gas of some neutral species, and the resulting ion-neutral complex both has an appreciable binding energy, and undergoes electron loss in this process, then an electron affinity for that complex can be derived from the scattering as a function of translational energy of the ion beam.
Non-Experimental Methods
Derivation [DER]
In a number of instances, authors have derived values for ionization energies or appearance energies through a variety of approaches. The most common type of derivation is based simply on taking the difference in enthalpies of formation of an ion and its corresponding neutral species, usually when for some reason an ionization energy can not be directly determined, but the necessary enthalpy of formation values are available from other measurements. This is not the only type of derivation appearing in the literature, however; results cited here include a variety of other types of derivation.
Estimation [EST]
In a few instances, results have been included that are designated as "estimated". The chief difference between a "derived" value and an "estimated" value is that derived values are usually closely tied to specific experimental results related to a particular ion, while "estimated" results usually come from an examination of trends in data for a series of molecules. Such results have been included here only when they come from studies that are sufficiently careful and systematic that the results appear to have some value for users searching for information about a particular ionic species. For both "derived" and "estimated" results, interested users should consult the original references for details.
Evaluation [EVAL]
Literature covered in putting together the database included reviews and papers where the authors have carried out expert evaluations of particular systems. Values recommended by the authors of such publications are included here and are specifically noted as "evaluations" by using the acronym EVAL.
Calculations [Calc]
The present work is intended to be an experimentally-based compilation. Although high level quantum calculations have been at times used in the evaluation of experimental data in the Webbook, they are not included in this work save for a very few cases where experimental formation of key ions in a series are unlikely to be successful.
Lattice Energy Calculations [Latt]
The heat of formation of an anion can be derived from a Born-Haber cycle using the lattice energy and heat of formation of a crystal and the thermochemistry of the appropriate gas phase cation. This method is not especially accurate relative to more recent techniques, but for some singly charged inorganic anions it provides the only data available.
RELIABILITY OF DATA AND CRITERIA FOR EVALUATION
Comparisons among Results of Different Techniques
The ion energetics data included in the WebBook are derived from the various different methods described above, and are consequently of widely varying quality, not only because the accuracies of the measurement techniques differ, but also because of differences in the focuses of the research in which the measurements were made. For example, many of the ionization energies reported for inorganic species were never intended by the original authors to be quantitative ionization energy measurement, but are simply qualitative indicators of whether or not a given ion observed in the vapor over a heated Knudsen cell has been formed by electron impact ionization of the corresponding neutral species (in which case it exhibits an onset at a relatively low energy) or through fragmentation of a molecular ion (which would correspond to a higher onset energy). In these experiments, error limits of ±0.5 to l eV are commonly cited by the original authors.
In carrying out an evaluation of data to arrive at a recommended value for an ionization energy, an attempt is made to integrate the entire corpus of information about any given ion, giving weight to various determinations depending on the nature of the ionization onset, the measurement techniques used, the attention to detail by the original authors, and so forth. For atoms or diatomic molecules, a spectroscopically-determined ionization energy is usually (but not always) considered more reliable than a contradictory value obtained by observation of an ionization threshold. Data obtained from coincidence experiments and other highly accurate measurements based on laser spectroscopy techniques are usually considered to be the most reliable. A value obtained from an observed ionization onset using photoionization or electron ionization with well-defined electron energies is considered more reliable than an onset obtained using less accurate techniques. In all of these cases, an observed onset of a photoelectron band is given great weight in carrying out the analysis, with values from simple onset observations being downgraded if they do not match the photoelectron onset (unless, of course, the differences could be rationalized in terms of the principles outlined above).
As mentioned above, many photoelectron spectroscopy studies do not cite values for adiabatic ionization energies. In cases where the authors have provided a figure showing the photoelectron spectrum, it is sometimes possible to estimate from the figure the value for the adiabatic onset; where listed adiabatic ionization energies have been obtained in this way, the values are shown surrounded by parentheses, and should be considered to be only rough estimates.
Most of the proton affinity data have been derived from ion/molecule equilibrium constant determinations. The values reported here are taken from the recent re-evaluation of proton affinity data [10], and differ somewhat from those listed in the frequently-cited 1984 evaluation [8], since the scale of relative values of proton affinities was shown to have been constricted, and has now been expanded in agreement with recent results and calculations [10]. Other equilibrium constant data have been utilized as an aid in evaluating information obtained from other sources.
Error Limits
The experimentally-determined data collected here display widely varying uncertainties, ranging from ±0.0001 eV or smaller for some optical- or laser-spectroscopic determinations to ±1 eV for electron ionization measurements carried out on the vapor above a heated Knudsen cell. The error limits shown for any particular determination are those given by the original author(s). When the original authors do not indicate error limits, no limits are listed here. Some of the ionization energy values are shown enclosed in parentheses. These indicate data which were not reported in the original paper, but which have been derived by us from a figure or other information in the paper.
In some cases, a recommended value for an ionization energy is given. Recommended values may originate from an analysis of the original experimental data listed for the species; in this case, cited error limits are derived from the various experimentally-determined values for the ionization energy using standard statistical analyses of the data. As explained above, different weights are given to determinations, depending on the accuracy of the experimental technique used; in most cases, this means that when measurements derived from reliable techniques are available, those that are clearly unreliable (for example, an ionization onset determination made with an electron beam where the energy spread of the electrons is broad) are disregarded. In a few cases, recommended values come from reviews in which the authors have carried out detailed data evaluations; when those evaluations are accepted for this database, the value reported in the review is listed, with "EVAL" (evaluation) given as the technique, and that value, with its error limits, is simply reproduced in the space for our recommended value.
REFERENCES
- F. H. Field and J. L. Franklin, Electron Impact Phenomena and the Properties of Gaseous Ions (Academic Press, New York, 1957).
- J. L. Franklin, J.G. Dillard, H. M. Rosenstock, J.T. Herron, K. Draxl, and F. H. Field, Ionization Potentials, Appearance Potentials and Heats of Formation of Gaseous Positive Ions , Nat. Stand. Ref. Data Ser., Nat. Bur. Stand. (U.S.) 26 (1969).
- H. M. Rosenstock, K. Draxl, B. W. Steiner, and J.T. Herron, Energetics of Gaseous Ions, J. Phys. Chem. Ref. Data, Vol. 6, Suppl. 1 (1977).
- R. D. Levin and S. G. Lias, Ionization Potential and Appearance Potential Measurements, 1971-1981, Nat. Stand. Ref. Data Ser., Nat. Bur. Stand. (U.S.) 71 (1982).
- (a) S. G. Lias and S. E. Stein, NIST Positive Ion Energetics, Versions 2.0 and 3.0, NIST Standard Reference Database 19A, National Institute of Standards and Technology, Gaithersburg, Maryland (1991 and 1993).
(b) J. E. Bartmess, NIST Negative Ion Energetics, Versions 2.0 and 3.0, NIST Standard Reference Database 19B, National Institute of Standards and Technology, Gaithersburg, Maryland (1991 and 1993). - (a) D. R. Stull and H. Prophet, JANAF Thermochemical Tables, Nat. Stand. Ref. Data Ser. (U.S.) 37 (1971).
(b) M. W. Chase, J. L. Curnutt, A. T. Hu, H. Prophet, A.N. Syverud, and L. C. Walker, JANAF Thermochemical Tables, 1974 Supplement, J. Phys. Chem. Ref. Data 3, 311 (1974).
(c) M. W. Chase, Jr., C. A. Davies, J. R. Downey, Jr., D. J. Frurip, R. A. McDonald, and A. N. Syverud, JANAF Thermochemical Tables, Third Edition, Parts I and II, J. Phys. Chem. Ref. Data 14, Suppl. 1 (1985).
(d) D. D. Wagman, W. H. Evans, V. B. Parker, R. H. Schumm, I. Halow, S. M. Bailey, K. L. Churney, and R. L. Nuttall, The NBS Tables of Chemical Thermodynamic Properties: Selected Values for Inorganic and C1 and C2 Organic Substances in SI Units, J. Phys. Chem. Ref. Data 11, Suppl. 2 (1982).
(e) L. V. Gurvich, I. V. Veits, V. A. Medvedev, G. A. Khachkuruzov, V. S. Yungman, G. A. Bergman, et al., Termodinamicheskie Svoistva Individual'nykh Veshchestv (Thermodynamic Properties of Individual Substances), V. P. Glushko, Gen. Ed., Vols. 1 through 4 (in 8 parts), (1978-1982), Izdatel'stvo "Nauka" Moscow. - S. G. Lias, J. E. Bartmess, J. F. Liebman, J. L. Holmes, R. D. Levin, and W. G. Mallard, "Gas-Phase Ion and Neutral Thermochemistry," J. Phys. Chem. Ref. Data, Vol. 16, Suppl. 1 (1988).
- S. G. Lias, J. F. Liebman, and R. D. Levin, Evaluated Gas Phase Basicities and Proton Affinities of Molecules; Heats of Formation of Protonated Molecules, J. Phys. Chem. Ref. Data, Vol. 13, 695 (1984).
- (a) S. G. Lias, J. F. Liebman, J. L. Holmes, R. D. Levin, and W. G. Mallard, NIST Positive Ion Energetics, Version 1.0, NIST Standard Reference Database 19A, National Institute of Standards and Technology, Gaithersburg, Maryland (1990). (b) J. E. Bartmess, NIST Negative Ion Energetics, Version 1.0, NIST Standard Reference Database 19B, National Institute of Standards and Technology, Gaithersburg, Maryland (1990).
- E. P. Hunter, and S. G. Lias, "Evaluated Gas Phase Basicities and Proton Affinities of Molecules", J. Phys. Chem. Ref. Data, Vol. 27(3), 413 (1998).
- E. R. Cohen, B. N. Taylor, J. Res. Nat. Bureau Stand., 92, 85 (1987).
- (a) J. E. Bartmess, J. Phys. Chem. 98, 6420 (1994).
(b) A. C. G. Mitchell, Z. Physik 51, 720 (1928). - H. M. Rosenstock, "Standard States in Gas Phase Ion Thermochemistry", in Kinetics of Ion-Molecule Reactions, (P. Ausloos, Editor), Plenum: New York, 1979, p. 246.
- H. M. Rosenstock, Int. J. Mass Spectrom. Ion Phys. 20, 139 (1976).
- D.S. McGrew, W.B. Knighton, J.A. Bognar, E.P. Grimsrud, Int. J. Mass Spectrom. Ion Phys. 139, 47 (1994); D.H. Williamson, W.B. Knighton, E.P. Grimsrud, Int. J. Mass Spectrom. Ion Phys. 154, 15 (1996).
- S. G. Lias and P. Ausloos, J. Am. Chem. Soc. 100, 6027 (1978).
- J. C. Traeger and R. G. McLaughlin, J. Am. Chem. Soc. 103, 3647 (1981).
- W. A. Brand and T. Baer, J. Am. Chem. Soc. 106, 3154 (1984).
- F. P. Lossing, Can. J. Chem. 50, 3973 (1972).
- (a) J. A. Simpson and C. E. Kuyatt, Rev. Sci. Instr. 34, 265 (1963).
(b) J. A. Simpson, Rev. Sci. Instru. 35, 1698 (1964).
(c) K. Maeda, G. P. Semeluk, and F. P. Lossing, J. Mass Spectrom. Ion. Phys. 1, 395 (1968).
(d) P. Marmet and L. Kerwin, Can. J. Phys. 38, 787 (1960). - (a) R. E. Fox, W. M. Kickam, T. Kjeldaas, Jr., and D. J. Grove, Phys. Rev. 84, 859 (1951).
(b) R. E. Fox, W. M. Hickam, D. J. Grove, and T. Kjeldaas, Jr., Rev. Sci. Instr. 26, 1101 (1955). - R. E. Winters, J. H. Collins, and W. L. Courchene, J. Chem. Phys. 45, 1931 (1966).
- J. Dannacher, Org. Mass Spectrom. 19, 253 (1984).
- R. D. Beck, P. Weis, G. Brauchle, and M. M. Kappes, Mechanistic aspects of fullerene coalescence upon ultraviolet laser desorption from thin films, J. Chem. Phys. 1994, 100, 262.
- R. S. Mullikan, J. Am. Chem. Soc. 74, 811 (1952).
- S. H. Hastings, J. L. Franklin, J. C. Schiller, and F. A. Matsen, J. Am. Chem. Soc. 75, 2900 (1953).
 |  | Standard Reference Data Program | Data Gateway | Chemistry WebBook |