
9.4: Biochemical nucleophilic substitutions with epoxide electrophiles
- Page ID
- 1093

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
9.4A: Hydrolysis of stearic acid epoxide: investigating the mechanism with kinetic isotope effect experiments
In chapter 8 we discussed some examples of nonenzymatic epoxide ring-opening reactions, and how they can proceed with either SN1-like or SN2-like mechanisms, depending on whether the reaction occurs in acidic or basic conditions. In a biological context, enzymes which catalyze the hydrolytic ring-opening of epoxides are called epoxide hydrolases. An epoxide hydrolase that has been isolated from soybeans, for example, is able to catalyze the hydrolysis of an epoxide derivative of stearate, a common fatty acid (J. Biol. Chem. 2005, 280, 6479).
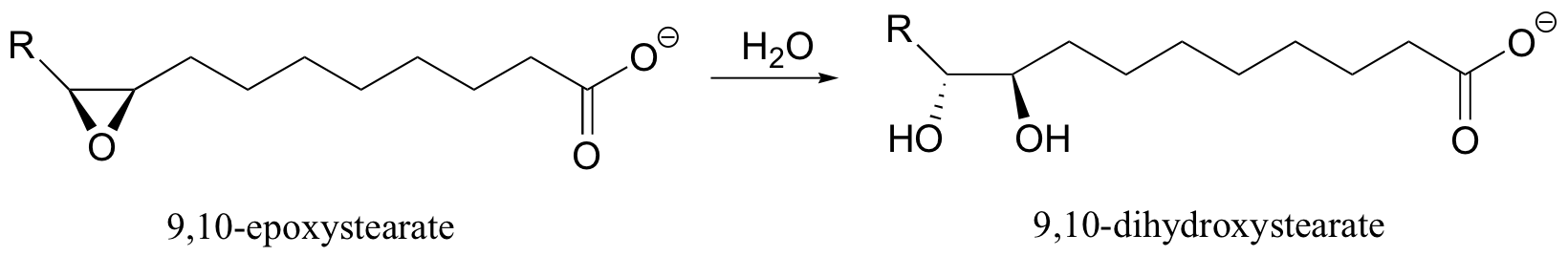
For a long time, scientists thought that epoxide hydrolase reactions involved direct nucleophilic attack by water. More recently, however, it has been determined that the ring-opening nucleophile is actually an aspartate in the enzyme's active site.
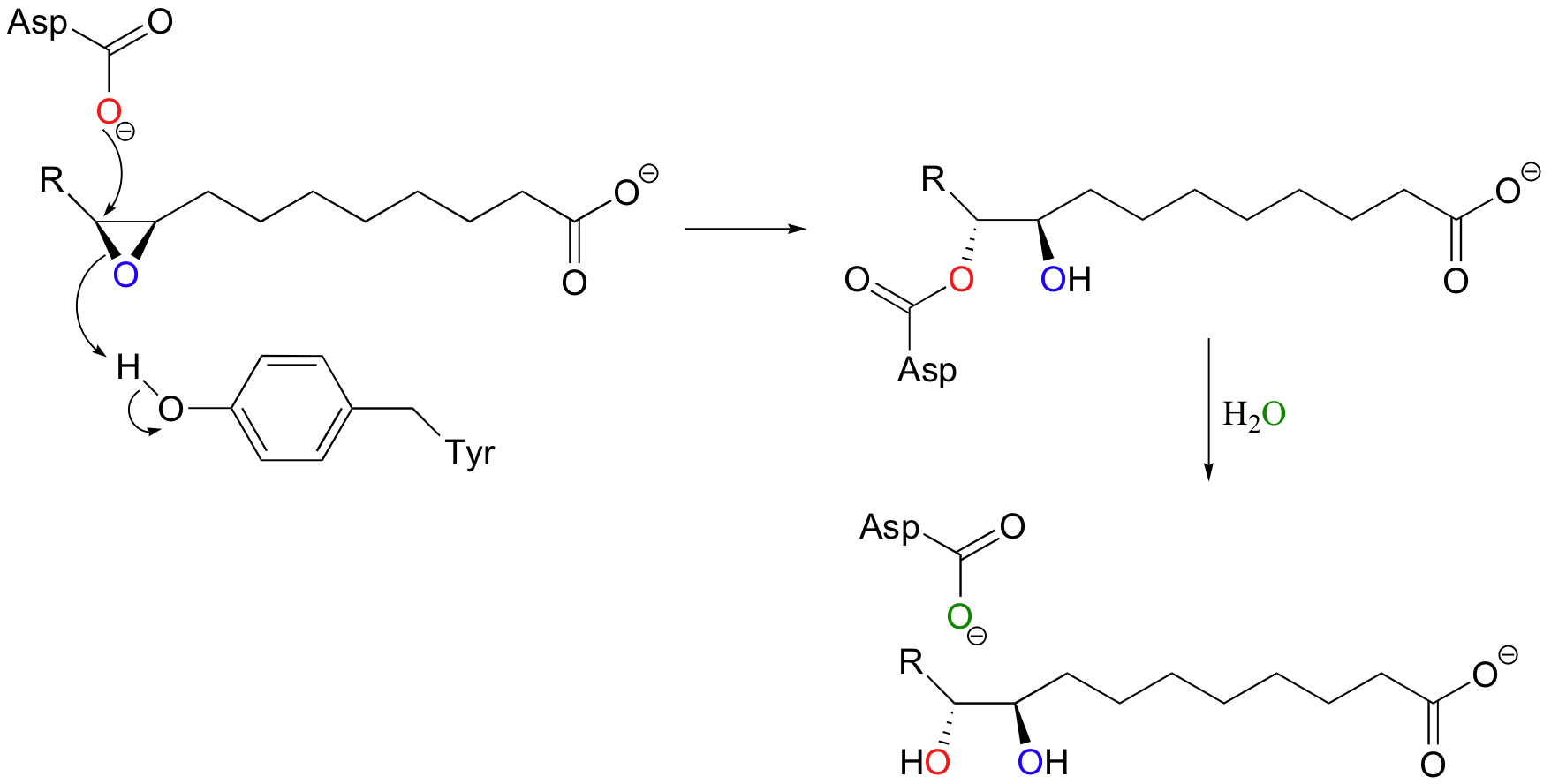
An active site acid (a tyrosine in this particular enzyme) protonates the epoxide oxygen, which lowers the energy barrier for ring-opening.
(You may be a little confused by what is happening in the second step, when the water molecule enters the picture. Don't worry about this yet - this type of reaction is the subject of chapter 12, and will make perfect sense when we get there. If you are looking at a color version of the figure above, however, you will notice from the color scheme that the oxygen in water does not end up in the diol product.)
Exercise 9.5: The second step of this reaction could be envisioned as a nucleophilic substitution, with water as the nucleophile and aspartate as the leaving group. In fact, this would be very similar to the mechanism of the retaining glycosidases covered previously. However, even if the oxygens in the figure above were not colored, the stereochemical outcome of the reaction should tell you that the second step is not an SN1 or SN2 reaction. Explain. (You will revisit this chemical step in a problem in chapter 12)
Solution
Let's focus on the first step, which is a nucleophilic substitution / epoxide ring opening reaction. Is this first step SN1-like or SN2-like? Researchers studying other epoxide hydrolase enzymes have presented evidence for a concerted, SN2-like mechanism. However, this particular plant enzyme appears to operate by a more SN1-like mechanism. What is the evidence that suggests this?
Previously, we discussed an experiment which used fluorine substituents on the reaction substrate to address this SN1 vs SN2 question. In this case, a very different approach was taken, using a phenomenon called the kinetic isotope effect.
When a hydrogen atom on a reacting molecule is replaced by a heavier deuterium or tritium isotope, small but measurable effects on the rate of the reaction are sometimes observed. These are called ‘kinetic isotope effects’ (KIE's). The term can also be applied to other isotopic substitutions such as 12C to 14C, but KIE's are most pronounced for hydrogen, because its heavy isotopes are two and three times heavier than 1H.
When the alpha-hydrogens of a ketone are replaced by deuterium, for example, the rate constant (k) for the acid-base reaction decreases by a factor of six (J. Am. Chem. Soc. 1972, 94, 8351).
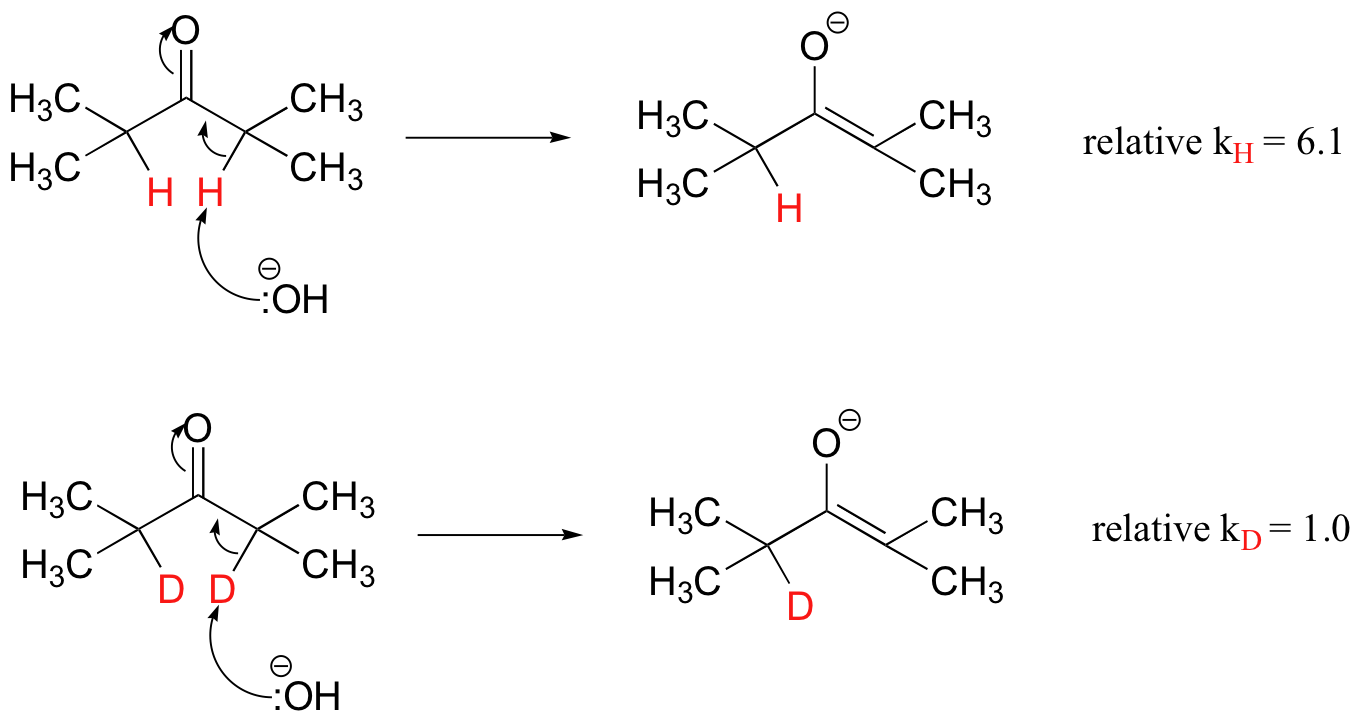
The KIE is expressed as the ratio of rate constants kH/kD (or kH/kT if tritium is used). In the above example, kH/kD = 6.1, meaning that replacement with deuterium slows down the reaction by a factor of 6.1 times. The reasoning behind the KIE effect is somewhat complex, but suffice it to say that the heavier mass of the deuterium makes the activation energy for the bond-breaking step higher. In the acid-base example above, the hydrogen/deuterium is directly involved in the bond-breaking event: it is being abstracted by a base. The observed effect is thus called a primary KIE.
A KIE is also often observed when the hydrogen isotope is not directly involved in the reaction, but rather is bound to a carbon that is involved in a bond-breaking or bond-forming step. This is called a secondary KIE, and an example is shown below (J. Am. Chem. Soc. 1970, 92, 232).
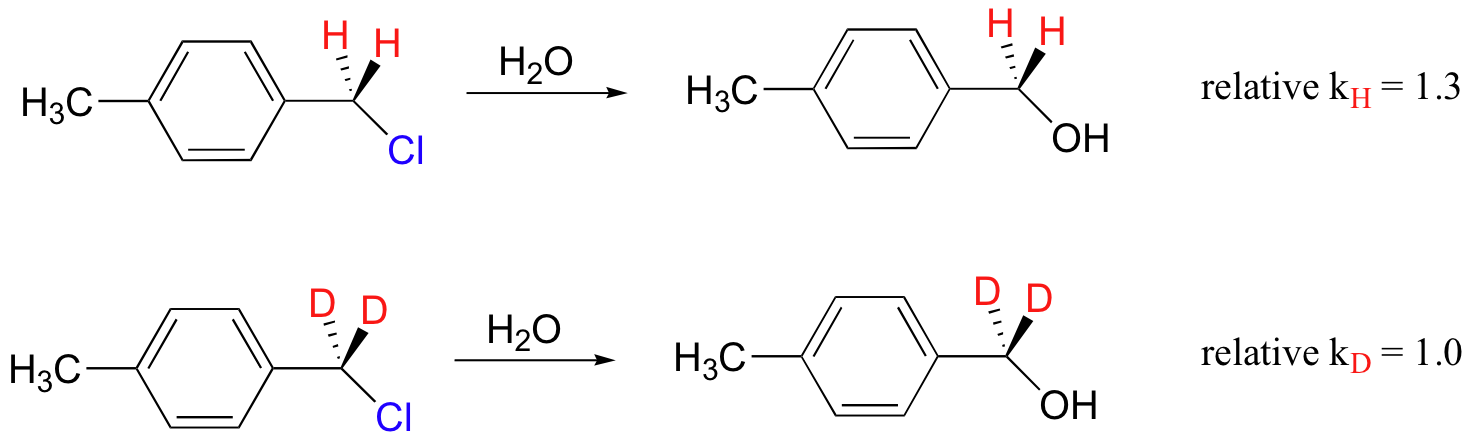
A secondary KIE can be either 'normal' (kH/kD > 1) or 'inverse' (kH/kD < 1, where the reaction with the heavier isotope actually goes faster).
The secondary KIE can be useful in determining reaction mechanism. In general, normal secondary KIE's are observed when the rate-determining step involves a carbon center changing from sp3 to sp2 hybridization, in particular when this step involves the formation of a carbocation. An SN1 reaction is expected to show a normal secondary KIE, because in the rate-determining step the carbon changes from sp3 neutral charge to sp2 positive charge. The reaction shown above, which is an SN1 hydrolysis, has a 'normal' kH/kD = 1.3. Conversely, if the rate-determining step involves a carbon center changing from sp2 to sp3 hybridization, an inverse secondary KIE is often observed (we will study this reaction type in chapter 11).
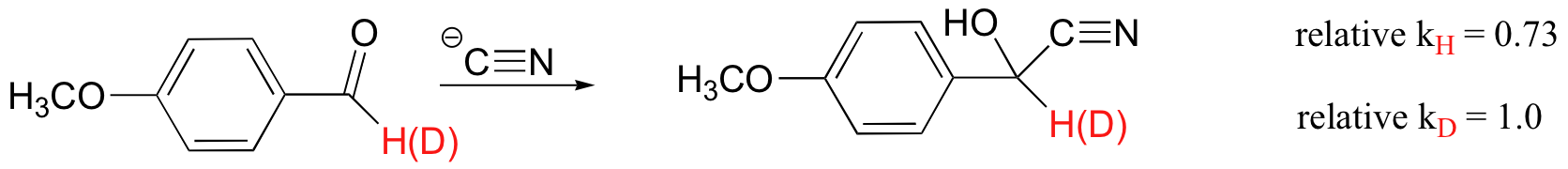
(J. Am. Chem. Soc. 1972, 94, 7579)
In SN2 substitutions, where there is no sp3 to sp2 change in hybridization and no carbocation intermediate, the KIE is generally either slightly inverse or close to unity (kH/kD = 1).
For the soybean epoxide hydrolase reaction described earlier, a normal secondary KIE of kH/kT = 1.30 was observed when a tritiated substrate was used (J. Biol. Chem. 2005, 280, 6479).
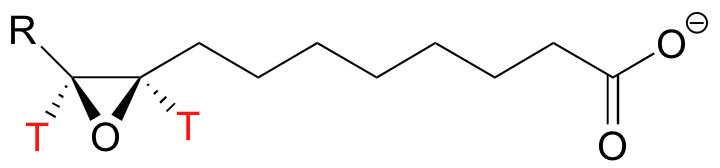
This indicates (but is not by any means conclusive proof of) a SN1-like mechanism for the ring-opening step of the reaction.
Note: The non-enzymatic KIE examples in this section were taken from Advanced Organic Chemistry by Francis A. Carey and Richard J. Sundberg, which is recommended as an excellent source for more information on the kinetic isotope effect (and many other topics in organic chemistry. Many more examples of how KIE experiments can be used to investigate enzyme mechanisms can be found in the references below:
1) Cook, P. F. (1991) in Enzyme Mechanism from Isotope Effects (Cook, P. F., ed) CRC Press, Boca Raton, FL
2) Schramm, V. L. Acc. Chem. Res. 2003 36, 588.
9.4B: Fosfomycin: an epoxide antibiotic
Fosfomycin is an antibiotic drug that acts by irreversibly inhibiting an enzyme involved in the biosynthesis of bacterial cell walls.
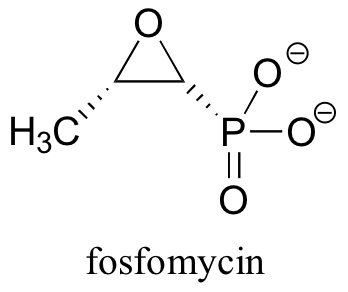
Specifically, fosfomycin shuts down the enzyme UDP-N-acetylglucosamine enolpyruvyl transferase, commonly known as MurA, which catalyzes a critical step in the construction of bacterial cell walls.
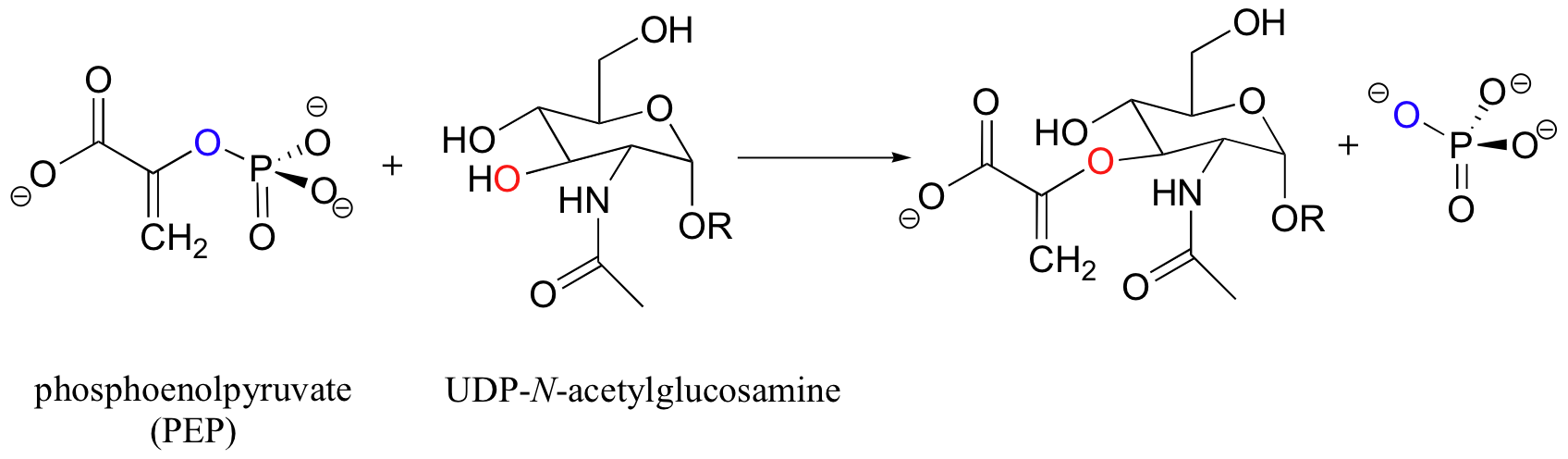
This may appear to be simply an SN1 or SN2 reaction - but recall (section 8.2E) that nucleophilic substitutions generally do not occur at alkene carbons! The enzymatic reaction itself is somewhat complex and outside the scope of the present discussion, but the important point for now is that an active site cysteine is known to be essential for the enzyme's catalytic function.
The key to the action of fosfomycin is its reactive epoxide group (Biochemistry 1994, 33, 10646), which serves as an electrophilic 'bait' for the active site cysteine.
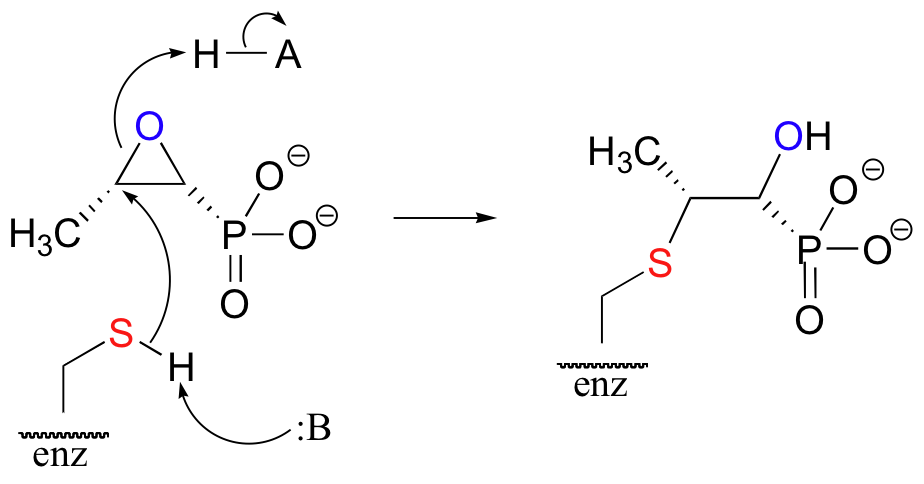
When the fosfomycin molecule (which is structurally a mimic of the phosphoenolpyruvate substrate) enters the active site of the enzyme, the cysteine thiol attacks one of the electrophilic carbons and opens the ring, leading to the irreversible alkylation of the active site. This effectively shuts down the enzyme, leading to death of the cell.
Exercise 9.6: Account for the regiochemical outcome of the enzyme-alkylating reaction shown above.