
13.6: Synthetic parallel - carbon nucleophiles in the lab
- Page ID
- 982

We've seen how enzymes use aldol and Claisen condensations to form new carbon-carbon bonds in the process of building large molecules. Chemists working in the laboratory can also carry out aldol and Claisen reactions, however they also have at their disposal a large and continuously growing toolbox of other carbon-carbon bond-forming reactions. In some respects, human chemists have an advantage, as they are able to work with conditions that are untenable in a living system: organic solvents, for example, or extremes of pH or temperature. Of course, enzymes have a distinct advantage when it comes to regio- and stereospecificity. Let's look at some commonly used laboratory procedures for creating new carbon-carbon bonds.
13.6A: Lab reactions with enolate /enamine intermediates
Aldol and Claisen-type reactions can be quite useful in the organic synthesis lab. In the acetoacetic ester synthesisreaction, ethyl acetoacetate (or another beta-ketoester) is the starting point for the construction of mono- or di-substituted ketones.
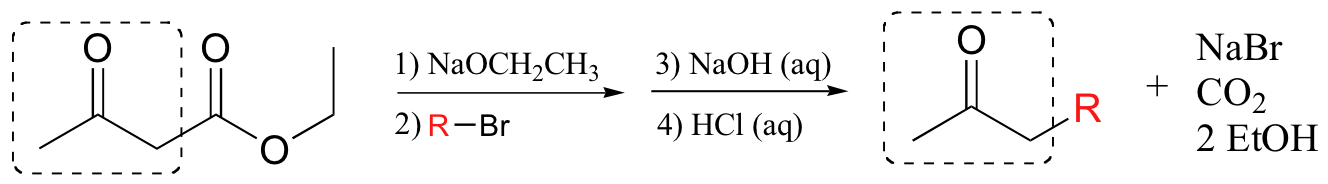
The key feature of beta-keto esters that makes them so useful for this purpose is the acidity of the ‘activated’ alpha-protons situated between the two carbonyl carbons – these protons typically have a pKa values in the range of 10-12, meaning that they can be efficiently deprotonated by alkoxide bases such as sodium ethoxide (step 1 above). The corresponding enolate nucleophile then attacks an alkyl halide in an SN2 reaction (step 2 - this is the carbon-carbon bond-forming step), then the ester is hydrolyzed with aqueous base (step 3) and the resulting beta-carboxylate group is acidified (step 4) and heated to induce decarboxylation. If we want to make a di-substituted ketone, we simply treat the monosubstituted product with a second alkyl halide before carrying out the hydrolysis and decarboxylation steps.
Exercise 13.12: Draw a complete mechanism for the following reaction:
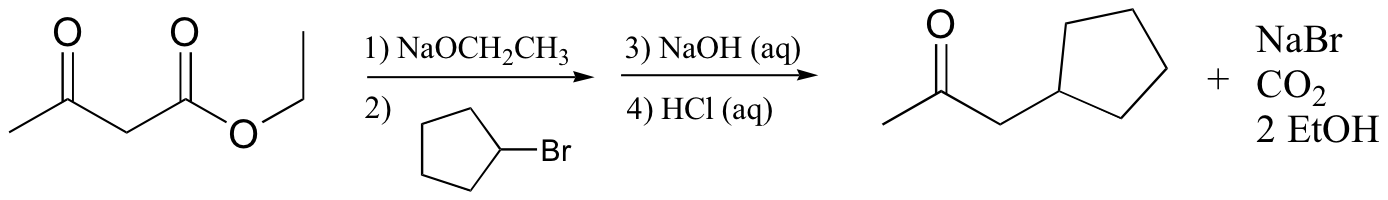
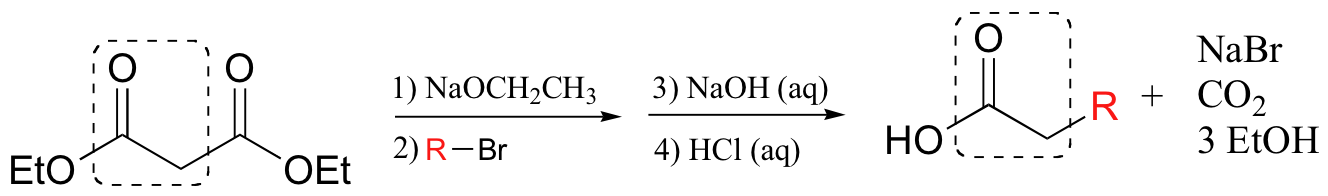
A similar process, commonly called the malonic ester synthesis, starts with a diester such as diethyl malonate and allows for the synthesis of substituted carboxylic acids.
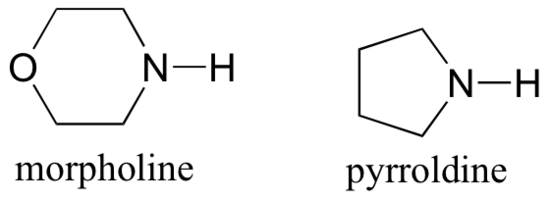
Stork enamine reactions closely parallel those catalyzed in living things by type I aldolases (section 13.3). In the Stork reaction, an alkyl or acyl group is added to a ketone or aldehdye via an enamine intermediate. While type I aldolases use the amino group of a lysine side chain to form an enamine, in a Stork reaction the synthetic chemist typically uses pyrrolidine or morpholine.
The examples below show the alkylation and acylation of acetone. For the alkylation reaction, a methyl or primary alkyl halide must be used.
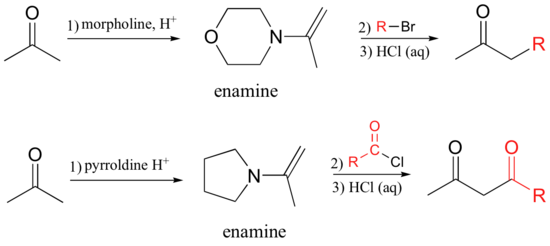
Exercise 13.13: Draw a complete mechanism for the Stork enamine acylation reaction shown above, assuming that the acylating agent is acetic acid chloride.
Why bother to form the Schiff base, rather than proceeding directly through an enolate intermediate? It turns out that the enamine intermediate is a better nucleophile than the enolate for a couple for reason. First, the nitrogen of an enamine is less likely to react as a nucleophile than the oxygen atom of an enolate – in other words, there is more C-alkylation under the Stork conditions. Second, because an enamine intermediate is a weaker base than an enolate, there is less chance of elimination reactions competing with the desired SN2 step (we’ll talk more about the competition between substitution and elimination in section 14.3).
In the haloform reaction, methyl ketones are fully halogenated in the presence of base:
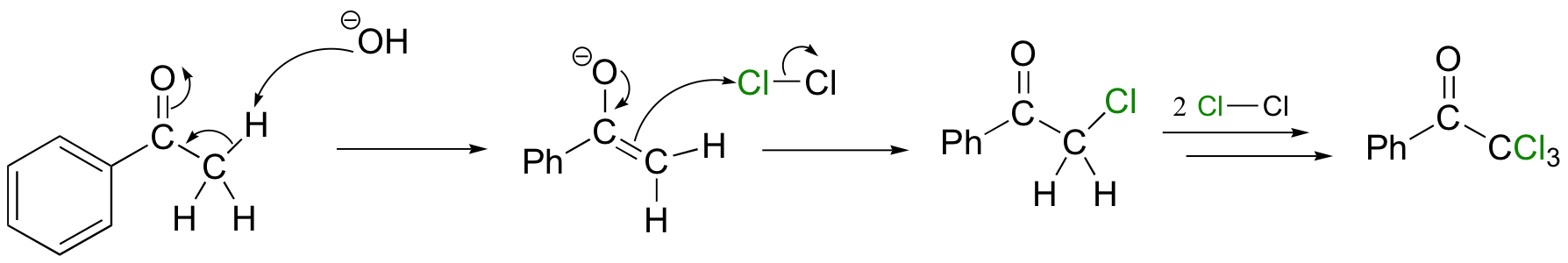
The electron-withdrawing effect of the three halogen substituents makes the tri-halomethyl anion a reasonable leaving group, which in turn allows for a somewhat unusual hydrolysis reaction and formation of the haloform and a carboxylic acid.

13.6B: The Wittig reaction
While aldol-type reactions are useful for carrying out many carbon-carbon bond-forming reactions in the lab, synthetic organic chemists are always trying to find new ways to create nucleophilic carbanion species. In the Wittig reaction, the negative charge on a carbanion is stabilized by a nearby positively charged phosphonium. Species such is this, in which positive and negative charges reside on adjacent atoms, are called ylides (we will encounter another kind of ylide species when we study some enzymatic reactions that depend on a form of thiamine, or vitamin B1, in section 14.5).
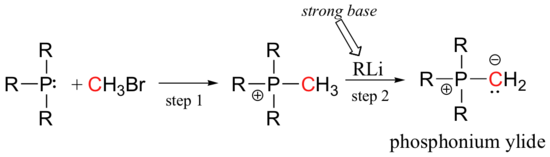
The carbanion next attacks a carbonyl carbon (step 3), after which bond rearrangement occurs (step 4).
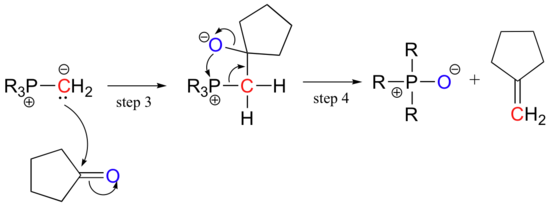
Notice that the overall result of a Wittig reaction is the formation of a double bond between a carbonyl carbon and a haloalkane.
Exercise13.14: Draw the structures of species A, B, and C in the reaction depicted below.
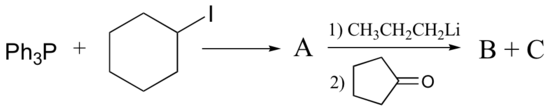
13.6C: Terminal alkynes as carbon nucleophiles
Recall from section 7.5 that the hydrogen on a terminal alkyne is somewhat acidic, with a pKa of approximately 26. This means that, given a strong enough base, terminal alkyne can be deprotonated, yielding a powerful carbanion nucleophile. Sodium amide in liquid ammonia is often used for this purpose.
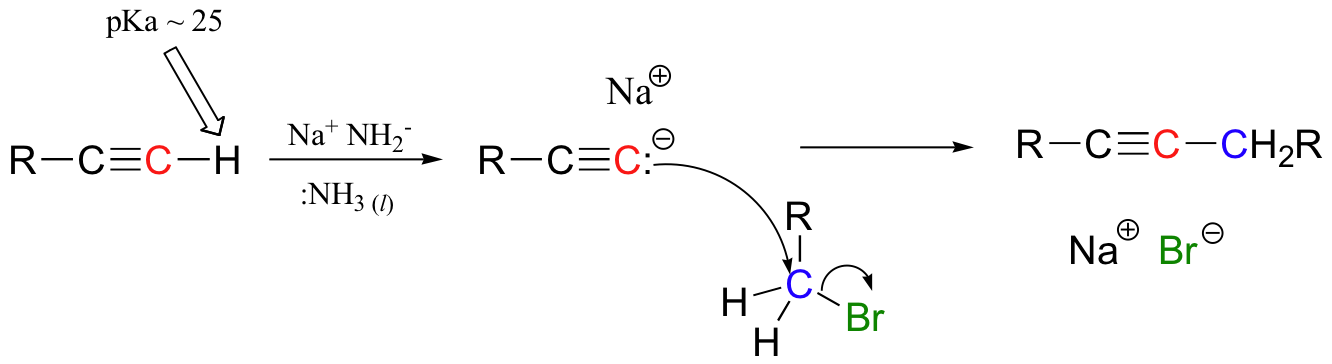
The alkynyl carbanion can then be combined with a suitable electrophile, such as a primary alkyl bromide, in a carbon-carbon bond-forming SN2 displacement reaction. We will discuss later in section 14.3A why secondary and tertiary alkyl bromides will usually not work in these types of reactions due to competition with elimination.
The alkyne group in the SN2 product can later be modified by other reactions: it can, for example, be reduced specifically to an E or Z alkene (these reactions are covered in section 16.5D).
When conducting a reaction such as this, we must take great care to avoid allowing the deprotonated alkyne, which of course is a powerful base as well as a nucleophile, to come into contact with water, alcohol, or anything else with an acidic proton. The result would be an acid-base reaction and regeneration of the original alkyne - not very productive! Reactions such as these are run in non-protic organic solvents such as diethyl ether or tetrahydrofuran, and all glassware used must be scrupulously dried. Generally a tube containing a drying agent such as calcium chloride is also affixed to the opening on the reaction flask to remove any moisture from the surrounding air.
13.6D: Grignard, Gilman, and organolithiuim reagents
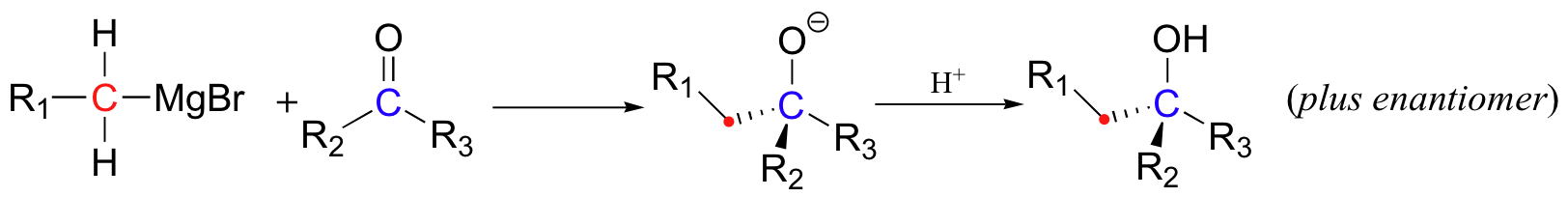
One of the most commonly used and versatile methods for forming a new carbon-carbon bond in the organic laboratory is a procedure called the 'Grignard reaction', named for the French chemist Victor Grignard (1871-1935) who won the 1912 Nobel Prize in Chemistry for developing this procedure. The carbon nucleophile is prepared by reacting an alkyl bromide or iodide with metallic magnesium, resulting in an organometallic compound in which the carbon reacts as if it were a carbanion.
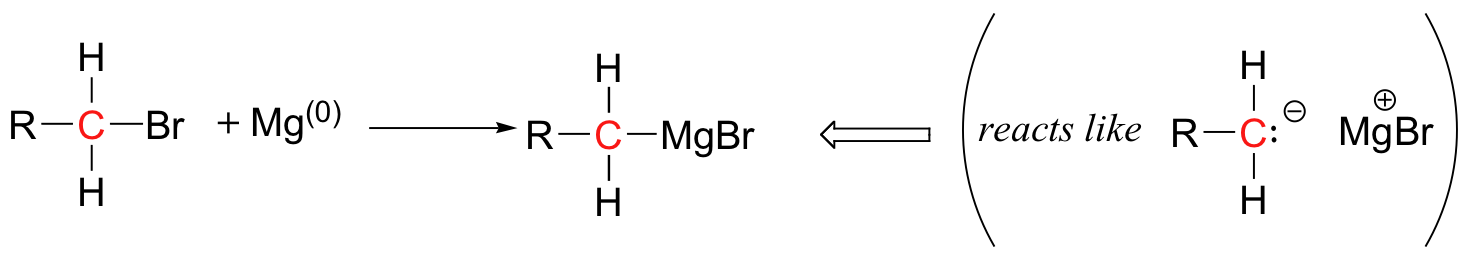
This 'Grignard reagent' is particularly useful in nucleophilic addition reactions with carbonyl electrophiles.
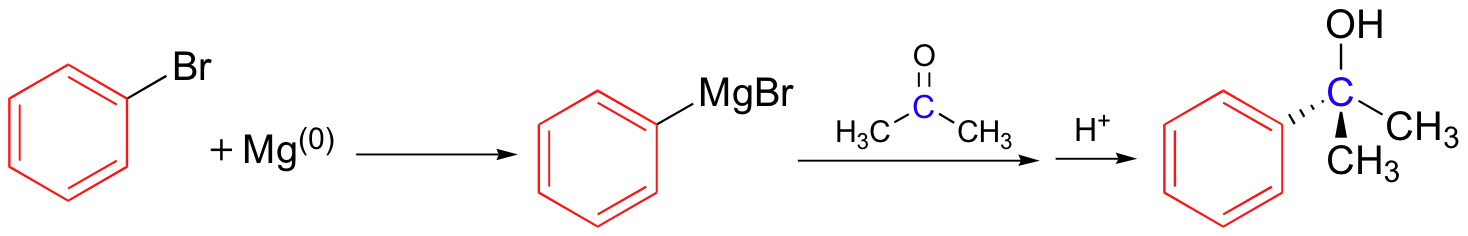
The nucleophilic carbon need not be primary as in the picture above. The synthesis of 2-phenyl-2-propanol, for example, can be carried by reacting phenyl magnesium bromide with acetone.
Grignard reagents can be also be carboxylated simply by pouring the organic solution over dry ice (solid CO2), then adding HCl:
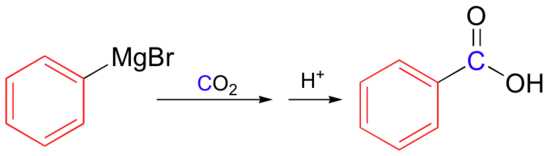
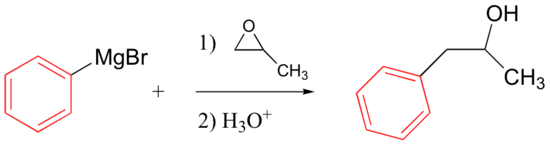
Grignard reagents will also react with epoxides, attacking the less hindered carbon (as expected for basic/nucleophilic ring-opening conditions – see section 8.6B).
The Grignard reagent is a very strong base, so the same precautions that were discussed above for alkynyl carbanions must be taken to avoid contact with water or anything that is even slightly acidic (in the syntheses above, acid is added only as the final step in order to recover protonated product and also to inactivate any remaining Grignard reagent). Because it is relatively simple to generate, a Grignard reagent such as phenyl magnesium bromide is also often used as the strong base (instead of sodium amide) in the deprotonation of terminal alkynes.
Esters and acid chlorides will react with two molar equivalents of Grignard reagent:
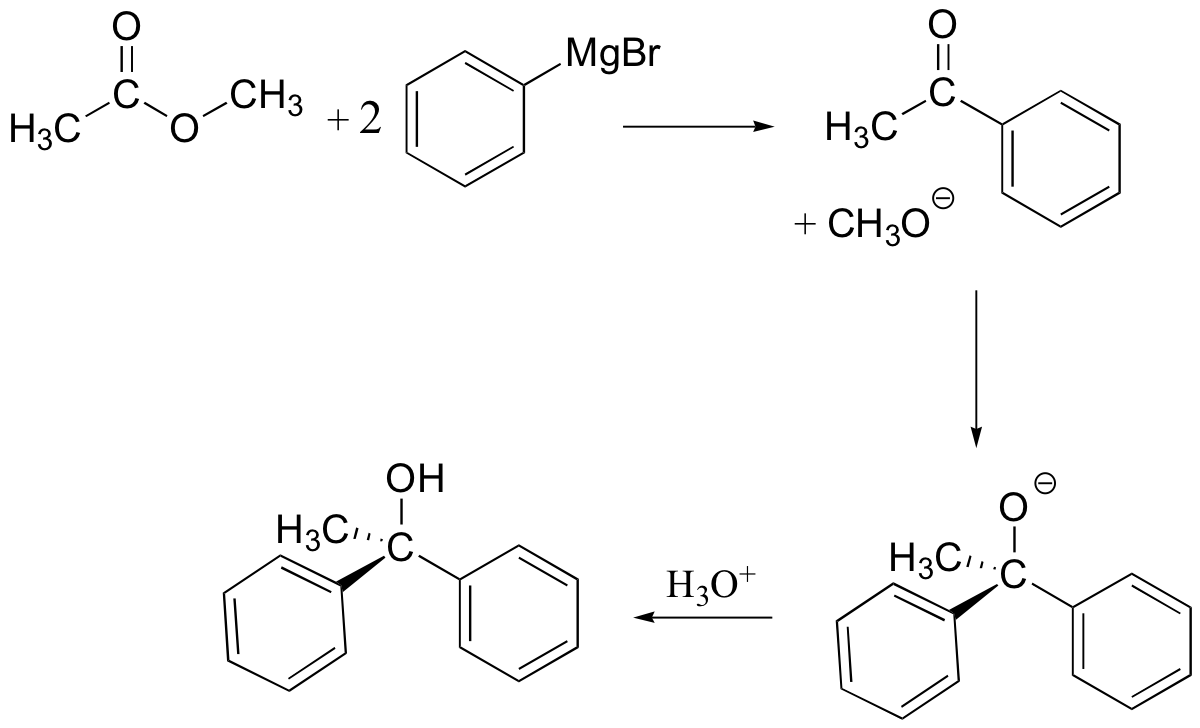
The first reaction is an acyl substitution reaction, and the second is a nucleophilic carbonyl addition. Because ketones and aldehydes are better electrophiles than carboxylic acid derivatives, it is not possible to stop these reactions at the ketone/aldehyde stage.
Carboxylic acids and amides will simply protonate the Grignard reagent, not a terribly productive reaction.
Grignard reagents will not react efficiently in SN2 reactions with alkyl halides (the Gilman reagent, described below, can be used for this purpose).
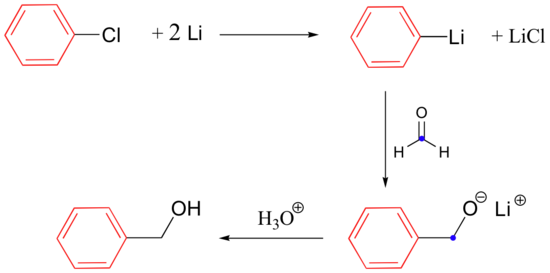
Metallic lithium can also be used to prepare organolithium compounds, which react in a fashion similar to Grignard reagents:
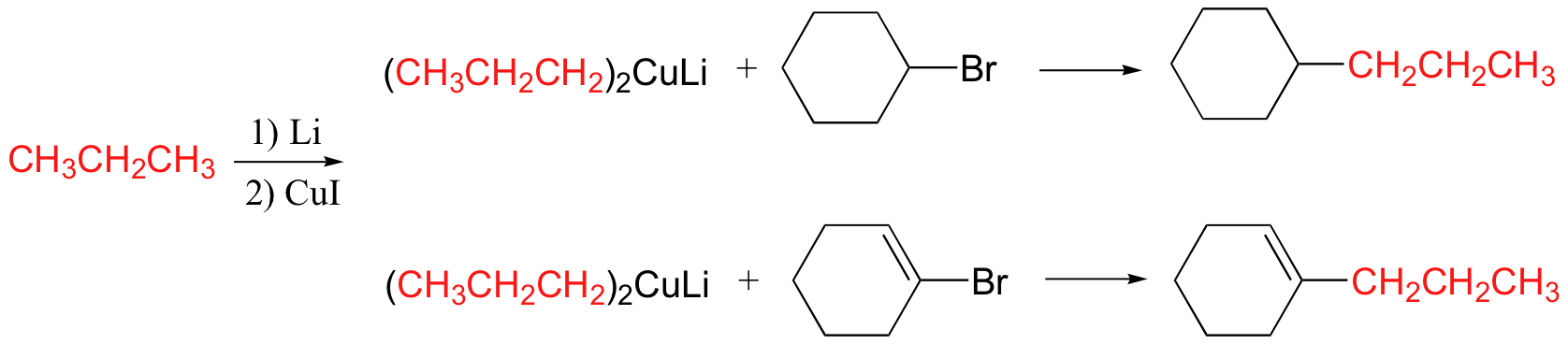
The Gilman reagent is a lithium diorganocopper species that can be prepared from organolithium compounds:

Gilman reagents are useful in that, unlike Grignard reagents, they will efficiently react in SN2 reactions with alkyl halides, even when the halogen is bonded to an sp2-hybridized, alkene carbon (remember from section 8.2C that SN2 reactions typically do not occur at sp2-hybridized carbons!)
Another useful property of Gilman reagent is that they can be used to add an alkyl group to an acid chloride just once (as opposed to twice, as is the case for Grignard reagents).
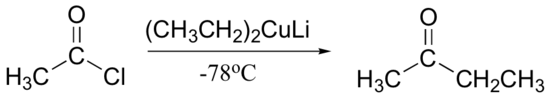
This reaction works only with acid chlorides - other carboxylic acid derivatives (esters, acid anhydrides, etc.) do not react with Gilman reagents.