
13.9: Physical Properties of Alcohols; Hydrogen Bonding
- Page ID
- 127213
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
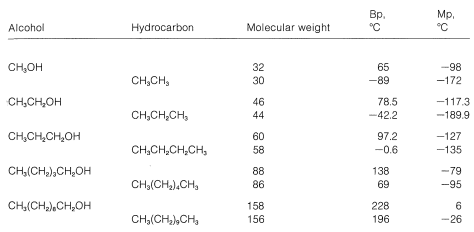
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)Comparison of the physical properties of alcohols with those of hydrocarbons of comparable molecular weight shows several striking differences, especially for those with just a few carbons. Alcohols are substantially less volatile, have higher melting points, and greater water solubility than the corresponding hydrocarbons (see Table 15-1), although the differences become progressively smaller as molecular weight increases.
Table 15-1: Comparison of Physical Properties of Alcohols and Hydrocarbons
The reason for these differences in physical properties is related to the high polarity of the hydroxyl group which, when substituted on a hydrocarbon chain, confers a measure of polar character to the molecule. As a result, there is a significant attraction of one molecule for another that is particularly pronounced in the solid and liquid states. This polar character leads to association of alcohol molecules through the rather positive hydrogen of one hydroxyl group with a correspondingly negative oxygen of another hydroxyl group:

This type of association is called “hydrogen bonding,” and, although the strengths of such bonds are much less than those of most conventional chemical bonds, they are still significant (about \(5\) to \(10 \: \text{kcal}\) per mole of hydrogen bonds). Clearly then, the reason alcohols have higher boiling points than corresponding alkyl halides, ethers, or hydrocarbons is because, for the molecules to vaporize, additional energy is required to break the hydrogen bonds. Alternatively, association through hydrogen bonds may be regarded as effectively raising the molecular weight, thereby reducing volatility (also see Section 1-3).
The water solubility of the lower-molecular-weight alcohols is pronounced and is understood readily as the result of hydrogen bonding with water molecules:

In methanol, the hydroxyl group accounts for almost half of the weight of the molecule, and it is not surprising that the substance is completely soluble in water. As the size of the hydrocarbon groups of alcohols increases, the hydroxyl group accounts for progressively less of the molecular weight, hence water solubility decreases (Figure 15-1). Indeed, the physical properties of higher-molecular-weight alcohols are very similar to those of the corresponding hydrocarbons (Table 15-1). The importance of hydrogen bonding in the solvation of ions was discussed in Section 8-7F.

Figure 15-1: Dependence of melting points, boiling points, and water solubilities of straight-chain primary alcohols \(\ce{H} \ce{-(CH_2)}_n \ce{-OH}\) on \(n\). The arrows on the solubility graph indicate that the scale is on the right ordinate.
Contributors
John D. Robert and Marjorie C. Caserio (1977) Basic Principles of Organic Chemistry, second edition. W. A. Benjamin, Inc. , Menlo Park, CA. ISBN 0-8053-8329-8. This content is copyrighted under the following conditions, "You are granted permission for individual, educational, research and non-commercial reproduction, distribution, display and performance of this work in any format."
Boiling Points
![]()
The chart below shows the boiling points of the following simple primary alcohols with up to 4 carbon atoms:
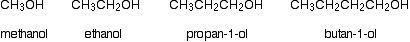
These boiling points are compared with those of the equivalent alkanes (methane to butane) with the same number of carbon atoms.
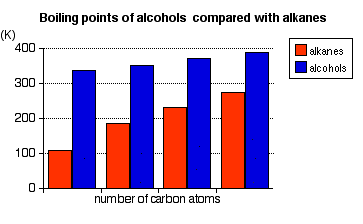
Notice that:
- The boiling point of an alcohol is always significantly higher than that of the analogous alkane.
- The boiling points of the alcohols increase as the number of carbon atoms increases.
The patterns in boiling point reflect the patterns in intermolecular attractions.
Hydrogen bonding
![]()
Hydrogen bonding occurs between molecules in which a hydrogen atom is attached to a strongly electronegative element: fluorine, oxygen or nitrogen. In the case of alcohols, hydrogen bonds occur between the partially-positive hydrogen atoms and lone pairs on oxygen atoms of other molecules.
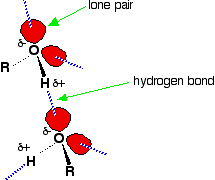
The hydrogen atoms are slightly positive because the bonding electrons are pulled toward the very electronegative oxygen atoms. In alkanes, the only intermolecular forces are van der Waals dispersion forces. Hydrogen bonds are much stronger than these, and therefore it takes more energy to separate alcohol molecules than it does to separate alkane molecules. This the main reason for higher boiling points in alcohols.
The effect of van der Waals forces
![]()
- Boiling points of the alcohols: Hydrogen bonding is not the only intermolecular force alcohols experience. There are also van der Waals dispersion forces and dipole-dipole interactions. The hydrogen bonding and dipole-dipole interactions are much the same for all alcohols, but dispersion forces increase as the alcohols get bigger. These attractions get stronger as the molecules get longer and have more electrons. This increases the sizes of the temporary dipoles formed. This is why the boiling points increase as the number of carbon atoms in the chains increases. It takes more energy to overcome the dispersion forces, and thus the boiling points rise.
- Comparison between alkanes and alcohols: Even without any hydrogen bonding or dipole-dipole interactions, the boiling point of the alcohol would be higher than the corresponding alkane with the same number of carbon atoms.
Compare ethane and ethanol:
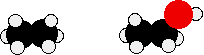
Ethanol is a longer molecule, and the oxygen atom brings with it an extra 8 electrons. Both of these increase the size of the van der Waals dispersion forces, and subsequently the boiling point. A more accurate measurement of the effect of the hydrogen bonding on boiling point would be a comparison of ethanol with propane rather than ethane. The lengths of the two molecules are more similar, and the number of electrons is exactly the same.
Solubility of alcohols in water
![]()
Small alcohols are completely soluble in water; mixing the two in any proportion generates a single solution. However, solubility decreases as the length of the hydrocarbon chain in the alcohol increases. At four carbon atoms and beyond, the decrease in solubility is noticeable; a two-layered substance may appear in a test tube when the two are mixed.
Consider ethanol as a typical small alcohol. In both pure water and pure ethanol the main intermolecular attractions are hydrogen bonds.
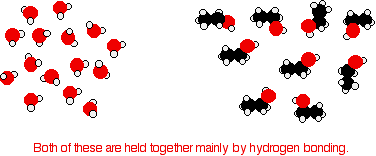
In order to mix the two, the hydrogen bonds between water molecules and the hydrogen bonds between ethanol molecules must be broken. Energy is required for both of these processes. However, when the molecules are mixed, new hydrogen bonds are formed between water molecules and ethanol molecules.
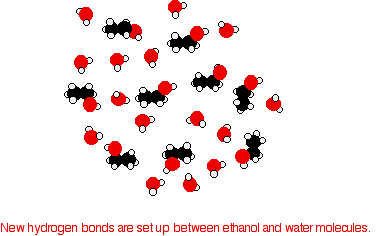
The energy released when these new hydrogen bonds form approximately compensates for the energy needed to break the original interactions. In addition, there is an increase in the disorder of the system, an increase in entropy. This is another factor in deciding whether chemical processes occur. Consider a hypothetical situation involving 5-carbon alcohol molecules.
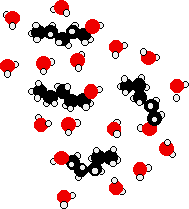
The hydrocarbon chains are forced between water molecules, breaking hydrogen bonds between those water molecules. The -OH ends of the alcohol molecules can form new hydrogen bonds with water molecules, but the hydrocarbon "tail" does not form hydrogen bonds. This means that many of the original hydrogen bonds being broken are never replaced by new ones.
In place of those original hydrogen bonds are merely van der Waals dispersion forces between the water and the hydrocarbon "tails." These attractions are much weaker, and unable to furnish enough energy to compensate for the broken hydrogen bonds. Even allowing for the increase in disorder, the process becomes less feasible. As the length of the alcohol increases, this situation becomes more pronounced, and thus the solubility decreases.
Acid/Base properties of alcohols
![]()
Several important chemical reactions of alcohols involving the O-H bond or oxygen-hydrogen bond only and leave the carbon-oxygen bond intact. An important example is salt formation with acids and bases. Alcohols, like water, are both weak bases and weak acids. The acid ionization constant (Ka) of ethanol is about 10~18, slightly less than that of water. Ethanol can be converted to its conjugate base by the conjugate base of a weaker acid such as ammonia {Ka — 10~35), or hydrogen (Ka ~ 10-38). It is convenient to employ sodium metal or sodium hydride, which react vigorously but controllably with alcohols:
The order of acidity of various liquid alcohols generally is water > primary > secondary > tertiary ROH. By this we mean that the equilibrium position for the proton-transfer reaction (Equation 15-1) lies more on the side of ROH and OHe as R is changed from primary to secondary to tertiary; therefore, tert-butyl alcohol is considered less acidic than ethanol:
ROH+OH−⇌RO−+HOH(1)(1)ROH+OH−⇌RO−+HOH
However, in the gas phase the order of acidity is reversed, and the equilibrium position for Equation 15-1 lies increasingly on the side of ROGas R is changed from primary to secondary to tertiary, terf-Butyl alcohol is therefore more acidic than ethanol in the gas phase. This seeming contradiction appears more reasonable when one considers what effect solvation (or the lack of it) has on equilibria expressed by Equation 15-1. In solution, the larger anions of alcohols, known as alkoxide ions, probably are less well solvated than the smaller ions, because fewer solvent molecules can be accommodated around the negatively charged oxygen in the larger ions:
Acidity of alcohols therefore decreases as the size of the conjugate base increases. However, “naked” gaseous ions are more stable the larger the associated R groups, probably because the larger R groups can stabilize the charge on the oxygen atom better than the smaller R groups. They do this by polarization of their bonding electrons, and the bigger the group, the more polarizable it is. (Also see Section 11-8A, which deals with the somewhat similar situation encountered with respect to the relative acidities of ethyne and water.)
Chemical Reactions of Alcohols involving the O-H bond of Compounds with Basic Properties
![]()
Alcohols are bases similar in strength to water and accept protons from strong acids. An example is the reaction of methanol with hydrogen bromide to give methyloxonium bromide, which is analogous to the formation of hydroxonium bromide with hydrogen bromide and water:
Acidity of Phenol
![]()
Compounds like alcohols and phenol which contain an -OH group attached to a hydrocarbon are very weak acids. Alcohols are so weakly acidic that, for normal lab purposes, their acidity can be virtually ignored. However, phenol is sufficiently acidic for it to have recognizably acidic properties - even if it is still a very weak acid. A hydrogen ion can break away from the -OH group and transfer to a base. For example, in solution in water:

Phenol is a very weak acid and the position of equilibrium lies well to the left. Phenol can lose a hydrogen ion because the phenoxide ion formed is stabilised to some extent. The negative charge on the oxygen atom is delocalised around the ring. The more stable the ion is, the more likely it is to form. One of the lone pairs on the oxygen atom overlaps with the delocalised electrons on the benzene ring.
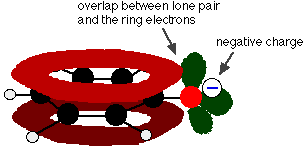
This overlap leads to a delocalization which extends from the ring out over the oxygen atom. As a result, the negative charge is no longer entirely localized on the oxygen, but is spread out around the whole ion.
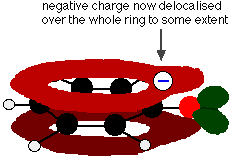
Spreading the charge around makes the ion more stable than it would be if all the charge remained on the oxygen. However, oxygen is the most electronegative element in the ion and the delocalized electrons will be drawn towards it. That means that there will still be a lot of charge around the oxygen which will tend to attract the hydrogen ion back again. That is why phenol is only a very weak acid.
Why is phenol a much stronger acid than cyclohexanol? To answer this question we must evaluate the manner in which an oxygen substituent interacts with the benzene ring. As noted in our earlier treatment of electrophilic aromatic substitution reactions, an oxygen substituent enhances the reactivity of the ring and favors electrophile attack at ortho and para sites. It was proposed that resonance delocalization of an oxygen non-bonded electron pair into the pi-electron system of the aromatic ring was responsible for this substituent effect. A similar set of resonance structures for the phenolate anion conjugate base appears below the phenol structures.
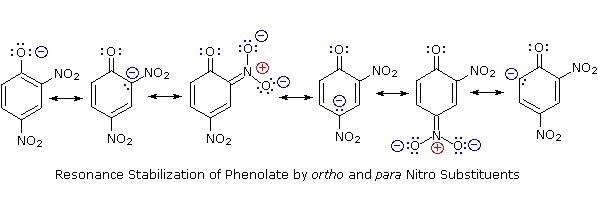
The resonance stabilization in these two cases is very different. An important principle of resonance is that charge separation diminishes the importance of canonical contributors to the resonance hybrid and reduces the overall stabilization. The contributing structures to the phenol hybrid all suffer charge separation, resulting in very modest stabilization of this compound. On the other hand, the phenolate anion is already charged, and the canonical contributors act to disperse the charge, resulting in a substantial stabilization of this species. The conjugate bases of simple alcohols are not stabilized by charge delocalization, so the acidity of these compounds is similar to that of water. An energy diagram showing the effect of resonance on cyclohexanol and phenol acidities is shown on the right. Since the resonance stabilization of the phenolate conjugate base is much greater than the stabilization of phenol itself, the acidity of phenol relative to cyclohexanol is increased. Supporting evidence that the phenolate negative charge is delocalized on the ortho and para carbons of the benzene ring comes from the influence of electron-withdrawing substituents at those sites.
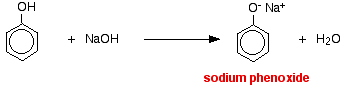
In this reaction, the hydrogen ion has been removed by the strongly basic hydroxide ion in the sodium hydroxide solution.
Acids react with the more reactive metals to give hydrogen gas. Phenol is no exception - the only difference is the slow reaction because phenol is such a weak acid. Phenol is warmed in a dry tube until it is molten, and a small piece of sodium added. There is some fizzing as hydrogen gas is given off. The mixture left in the tube will contain sodium phenoxide.
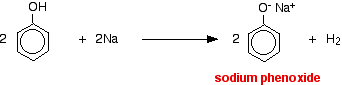
Acidity of Substituted Phenols
![]()
Substitution of the hydroxyl hydrogen atom is even more facile with phenols, which are roughly a million times more acidic than equivalent alcohols. This phenolic acidity is further enhanced by electron-withdrawing substituents ortho and para to the hydroxyl group, as displayed in the following diagram. The alcohol cyclohexanol is shown for reference at the top left. It is noteworthy that the influence of a nitro substituent is over ten times stronger in the para-location than it is meta, despite the fact that the latter position is closer to the hydroxyl group. Furthermore additional nitro groups have an additive influence if they are positioned in ortho or para locations. The trinitro compound shown at the lower right is a very strong acid called picric acid.
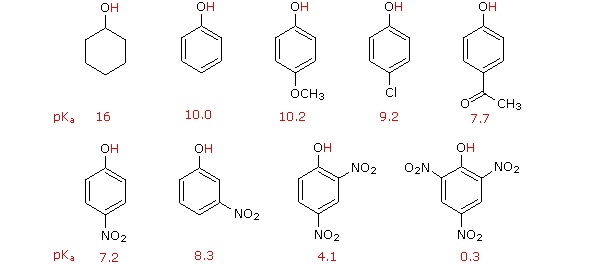
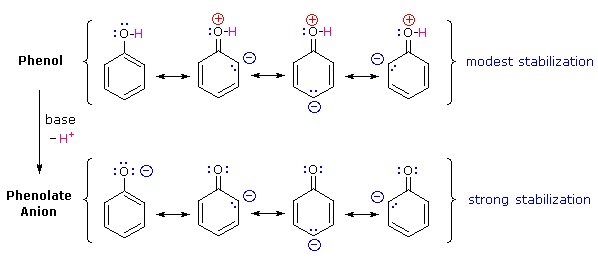
Why is phenol a much stronger acid than cyclohexanol? To answer this question we must evaluate the manner in which an oxygen substituent interacts with the benzene ring. As noted in our earlier treatment of electrophilic aromatic substitution reactions, an oxygen substituent enhances the reactivity of the ring and favors electrophile attack at ortho and para sites. It was proposed that resonance delocalization of an oxygen non-bonded electron pair into the pi-electron system of the aromatic ring was responsible for this substituent effect. Formulas illustrating this electron delocalization will be displayed when the "Resonance Structures" button beneath the previous diagram is clicked. A similar set of resonance structures for the phenolate anion conjugate base appears below the phenol structures.

The resonance stabilization in these two cases is very different. An important principle of resonance is that charge separation diminishes the importance of canonical contributors to the resonance hybrid and reduces the overall stabilization. The contributing structures to the phenol hybrid all suffer charge separation, resulting in very modest stabilization of this compound. On the other hand, the phenolate anion is already charged, and the canonical contributors act to disperse the charge, resulting in a substantial stabilization of this species. The conjugate bases of simple alcohols are not stabilized by charge delocalization, so the acidity of these compounds is similar to that of water. An energy diagram showing the effect of resonance on cyclohexanol and phenol acidities is shown on the right. Since the resonance stabilization of the phenolate conjugate base is much greater than the stabilization of phenol itself, the acidity of phenol relative to cyclohexanol is increased. Supporting evidence that the phenolate negative charge is delocalized on the ortho and para carbons of the benzene ring comes from the influence of electron-withdrawing substituents at those sites.
Contributors
![]()
-
Dr. Dietmar Kennepohl FCIC (Professor of Chemistry, Athabasca University)
-
Prof. Steven Farmer (Sonoma State University)
-
William Reusch, Professor Emeritus (Michigan State U.), Virtual Textbook of Organic Chemistry
-
Jim Clark (Chemguide.co.uk)
- John D. Robert and Marjorie C. Caserio (1977) Basic Principles of Organic Chemistry, second edition. W. A. Benjamin, Inc. , Menlo Park, CA. ISBN 0-8053-8329-8. This content is copyrighted under the following conditions, "You are granted permission for individual, educational, research and non-commercial reproduction, distribution, display and performance of this work in any format."