
20.16 Synthesis using Carbonyl Chemistry
- Page ID
- 28393

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)An Introduction to Synthesis
The study of organic chemistry exposes a student to a wide range of interrelated reactions. Alkenes, for example, may be converted to structurally similar alkanes, alcohols, alkyl halides, epoxides, glycols and boranes; cleaved to smaller aldehydes, ketones and carboxylic acids; and enlarged by carbocation and radical additions as well as cycloadditions. All of these products may be transformed subsequently to a host of new compounds incorporating a wide variety of functional groups, and thereby open to even further elaboration. Consequently, the logical conception of a multistep synthesis for the construction of a designated compound from a specified starting material becomes one of the most challenging problems that may be posed.
A one or two step sequence of simple reactions is not that difficult to deduce. If, for example, one is asked to prepare meso-3,4-hexanediol from 3-hexyne, most students realize it will be necessary to reduce the alkyne to cis or trans-3-hexene before undertaking glycol formation. Permanganate or osmium tetroxide hydroxylation of cis-3-hexene would form the desired meso isomer. From trans-3-hexene it would be necessary to first epoxidize the alkene with a peroxyacid, followed by ring opening with hydroxide ion. This example illustrates a common feature in synthesis: often there is more than one effective procedure that leads to the desired product.
Longer multistep syntheses require careful analysis and thought, since many options need to be considered. Like an expert chess player evaluating the long range pros and cons of potential moves, the chemist must appraise the potential success of various possible reaction paths, focussing on the scope and limitations constraining each of the individual reactions being employed. This can be a daunting task, the skill for which is acquired by experience, and often trial and error.
The three examples shown below are illustrative. The first is a simple functional group conversion problem, that may initially seem difficult. It is often helpful to work such problems backwards, starting from the product. In this case it should be apparent that cyclohexanol may be substituted for cyclohexanone, since the latter could then be made by a simple oxidation. Also, since cyclohexane (and alkanes in general) is relatively unreactive, bromination (or chlorination) would seem to be an obvious first step. At this point one is tempted to convert bromocyclohexane to cyclohexanol by an SN2 reaction with hydroxide ion. This reaction would undoubtedly be accompanied by E2 elimination, so it would be cleaner, although one step longer, to first make cyclohexene and then hydrate it by any of several methods (e.g. oxymercuration and hydroboration) including the one shown.
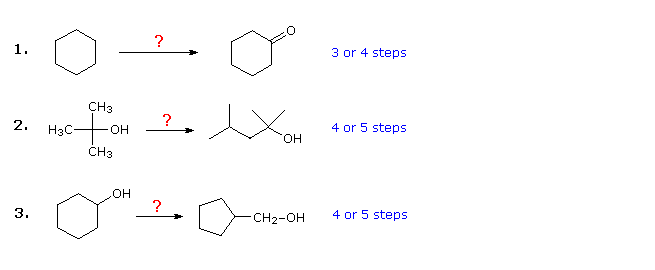
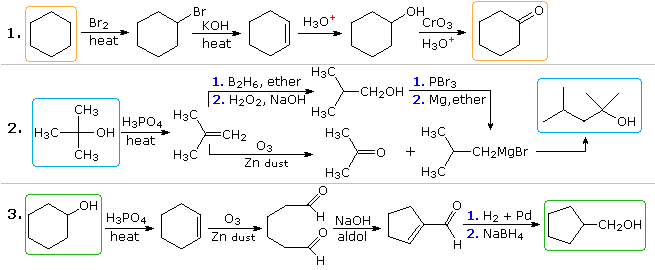
In problem 2 the desired product has seven carbon atoms and the starting material has four. Clearly, two intermediates derived from the starting compound must be joined together, and one carbon must be lost, either before or after this bonding takes place. The 3º-alcohol function in the product suggests formation by a Grignard addition to a ketone, and isobutene appears to be a good precursor to each of these reactants, as shown. The reactant and product compounds in the third problem are isomers, but some kind of bond-breaking and bond-making sequence is clearly necessary for this structural change to occur. One possible procedure is shown above. Acid-catalyzed rearrangement of cyclohexene oxide, followed by reduction might also serve.
Retrosynthetic Analysis
The useful approach of working out syntheses starting from the target molecule and working backward toward simpler starting materials has been formalized by Prof. E. J. Corey (Harvard) and termed retrosynthetic analysis. In this procedure the target molecule is transformed progressively into simpler structures by disconnecting selected carbon-carbon bonds. These disconnections rest on transforms, which are the reverse of plausible synthetic constructions. Each simpler structure, so generated, becomes the starting point for further disconnections, leading to a branched set of interrelated intermediates. A retrosynthetic transform is depicted by the => symbol, as shown below for previous examples 2 & 3. Once a complete analysis has been conducted, the desired synthesis may be carried out by application of the reactions underlying the transforms.

The above diagram does not provide a complete set of transforms for these target compounds. When a starting material is specified, as in the above problems, the proposed pathways must reflect that constraint. Thus the 4-methyl-2-pentanone and 3-methylbutyrate ester options in example 2, while entirely reasonable, do not fit well with a tert-butanol start. Likewise, a cyclopentyl intermediate might provide an excellent route to the product in example 3, but does not meet the specified conditions of the problem.
Retrosynthetic analysis is especially useful when considering relatively complex molecules without starting material constraints. If it is conducted without bias, unusual and intriguing possibilities sometimes appear. Unfortunately, molecular complexity (composed of size, functionality, heteroatom incorporation, cyclic connectivity and stereoisomerism) generally leads to very large and extensively branched transform trees. Computer assisted analysis has proven helpful, but in the end the instincts and experience of the chemist play a critical role in arriving at a successful synthetic plan. Some relatively simple examples, most having starting material restrictions, are provided below.
Examples
Problem 1
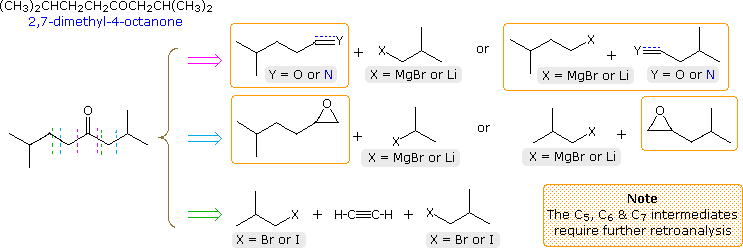
A synthesis of 2,7-dimethyl-4-octanone from starting compounds having no more than four contiguous carbon atoms is required. The structural formula and a first-stage retroanalysis of this ketone are displayed in the following diagram. Three straightforward disconnections are shown, as drawn by the dashed lines. The first (magenta arrow) is undoubtedly the simplest, since a Grignard reagent addition to a suitable nitrile gives the product directly. However, one or more of the reactants is larger than C4 and must therefore be prepared independently before use. A two-step procedure involving Grignard addition to an aldehyde, followed by oxidation of the 2º-alcohol product, also suffers the same requirement, as do the epoxide opening routes presented in the second row (cyan arrow). Secondary preparations of these intermediates are easily conceived by way of cyanide substitution of a 1º-halide, coupling of a Gilman reagent with allyl bromide, or Grignard addition to ethylene oxide.
The last disconnection (green arrow) creates the desired carbon skeleton by sequential alkylations of terminal alkynes (first acetylene and then 4-methyl-1-pentyne). Mercury catalyzed hydration of the symmetrical octyne product generates the desired ketone. All the necessary reactants are C4 or less, so the synthesis is accomplished in three steps (not counting the formation of alkyne salts).
Three more first-stage analyses will be displayed below. The first of these (red arrow) is a two step sequence initiated by isobutyl magnesium bromide addition to acetonitrile, followed by isobutyl bromide alkylation of the resulting 4-methyl-2-pentanone. Regioselective control might be a problem in the last step. The second disconnection (orange arrow) suggests an α, α'-dialkylation of acetone. Since acetone itself is prone to base-catalyzed condensation, this might be difficult to accomplish directly. However, the use of ethyl acetoacetate avoids this problem for the first step, and the second alkylation is the same one proposed as part of the first disconnection synthesis. Both of these sequences would provide efficient routes to the target ketone.
Finally, the last disconnection is a four component assembly consisting of two conjugate additions and a Grignard addition. This would most likely result in a longer and lower yield procedure than the previous two.
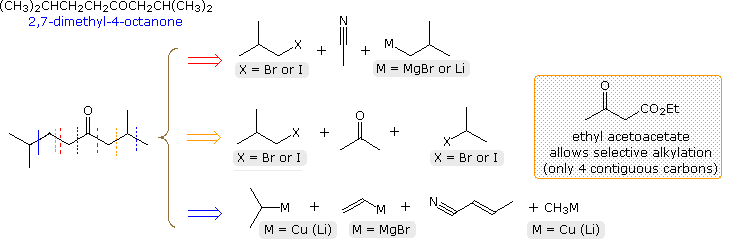
Problem 2
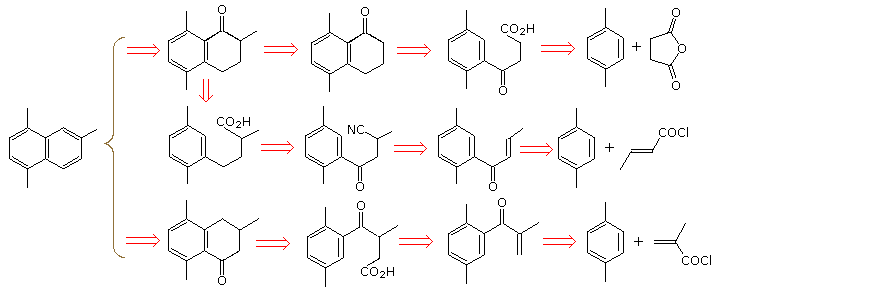
A synthesis of 1,4,6--trimethylnaphthalene from para-xylene and other starting compounds having no more than four contiguous carbon atoms is required. Plausible transforms for the attachment of the second ring carbons to para-xylene are Friedel-Craft alkylation or acylation (acylation is usually better), nucleophilic attack of an aryl metal reagent derived from 2-bromo-para-xylene on carbonyl or epoxide electrophiles, or possibly by cycloaddition to a aryne intermediate. A palladium catalyzed coupling reaction might also prove useful. Because of their simplicity and broad scope, we shall consider only the first two transforms.
The following diagram shows retrosynthetic analyses based on the Friedel-Craft transform for both bond formations to the aromatic ring. Of these, the first seems to offer the most efficient synthesis route, consisting of Friedel-Craft acylation, Wolff-Kischner reduction, a second Friedel-Craft acylation and methylation of a ketone enolate. In all cases the substituted tetralone precursor of the desired naphthalene must be reduced to an alcohol and dehydrated. The resulting dihydro naphthalene is then aromatized by Pt catalyzed dehydrogenation, or mild oxidation by heating with sulfur or selenium.
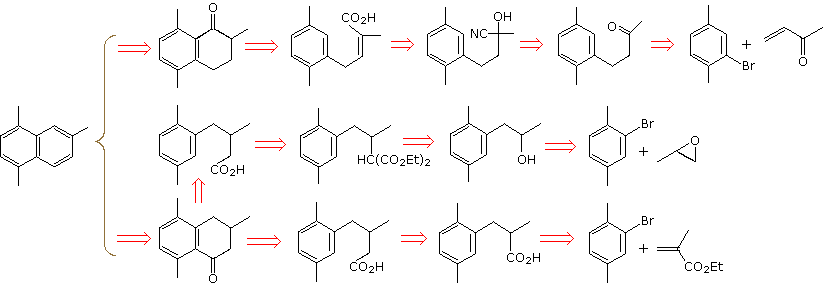
A new set of disconnections, starting from 2-bromo-para-xylene, will be displayed. A derived Gilman or lithium reagent is used for conjugate addition to an unsaturated carbonyl compound or ring opening of an epoxide. Further lengthening of the side chain is effected by cyanohydrin formation (top example), malonic ester alkylation (middle example), and Arndt-Eistert homologation (bottom example). The final steps must then parallel those used for the first examples.
Contributors
William Reusch, Professor Emeritus (Michigan State U.), Virtual Textbook of Organic Chemistry