
19.9 Acidity of Carboxylic Acids and Phenols
- Page ID
- 28371

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)Comparing the strengths of weak acids
The strengths of weak acids are measured on the pKa scale. The smaller the number on this scale, the stronger the acid is. Three of the compounds we shall be looking at, together with their pKa values are:
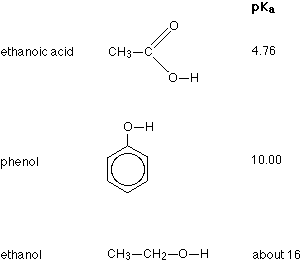
Remember - the smaller the number the stronger the acid. Comparing the other two to ethanoic acid, you will see that phenol is very much weaker with a pKa of 10.00, and ethanol is so weak with a pKa of about 16 that it hardly counts as acidic at all!
Acidity of Carboxylic Acids
The pKa 's of some typical carboxylic acids are listed in the following table. When we compare these values with those of comparable alcohols, such as ethanol (pKa = 16) and 2-methyl-2-propanol (pKa = 19), it is clear that carboxylic acids are stronger acids by over ten powers of ten! Furthermore, electronegative substituents near the carboxyl group act to increase the acidity.
|
Compound |
pKa |
Compound |
pKa |
|
|---|---|---|---|---|
| HCO2H | 3.75 | CH3CH2CH2CO2H | 4.82 | |
| CH3CO2H | 4.74 | ClCH2CH2CH2CO2H | 4.53 | |
| FCH2CO2H | 2.65 | CH3CHClCH2CO2H | 4.05 | |
| ClCH2CO2H | 2.85 | CH3CH2CHClCO2H | 2.89 | |
| BrCH2CO2H | 2.90 | C6H5CO2H | 4.20 | |
| ICH2CO2H | 3.10 | p-O2NC6H4CO2H | 3.45 | |
| Cl3CCO2H | 0.77 | p-CH3OC6H4CO2H | 4.45 |
Why should the presence of a carbonyl group adjacent to a hydroxyl group have such a profound effect on the acidity of the hydroxyl proton? To answer this question we must return to the nature of acid-base equilibria and the definition of pKa , illustrated by the general equations given below. These relationships were described in an previous section of this text.
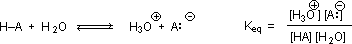
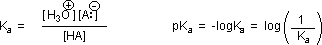
We know that an equilibrium favors the thermodynamically more stable side, and that the magnitude of the equilibrium constant reflects the energy difference between the components of each side. In an acid base equilibrium the equilibrium always favors the weaker acid and base (these are the more stable components). Water is the standard base used for pKa measurements; consequently, anything that stabilizes the conjugate base (A:(–)) of an acid will necessarily make that acid (H–A) stronger and shift the equilibrium to the right. Both the carboxyl group and the carboxylate anion are stabilized by resonance, but the stabilization of the anion is much greater than that of the neutral function, as shown in the following diagram. In the carboxylate anion the two contributing structures have equal weight in the hybrid, and the C–O bonds are of equal length (between a double and a single bond). This stabilization leads to a markedly increased acidity, as illustrated by the energy diagram.
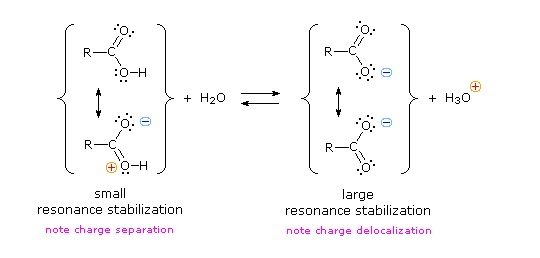
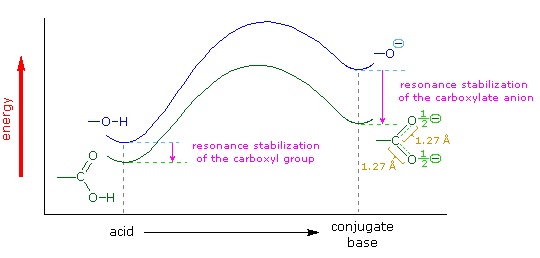
The pKa of ethanol is about 17, while the pKa of acetic acid is about 5: this is a 1012-fold difference in the two acidity constants. In both compounds, the acidic proton is bonded to an oxygen atom. How can they be so different in terms of acidity? We begin by considering the conjugate bases.
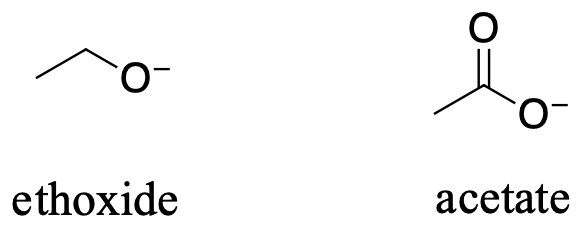
In both species, the negative charge on the conjugate base is held by an oxygen, so periodic trends cannot be invoked. For acetic acid, however, there is a key difference: a resonance contributor can be drawn in which the negative charge is localized on the second oxygen of the group. The two resonance forms for the conjugate base are equal in energy. What this means is that the negative charge on the acetate ion is not located on one oxygen or the other: rather it is shared between the two. Chemists use the term ‘delocalization of charge’ to describe this situation. In the ethoxide ion, by contrast, the negative charge is ‘locked’ on the single oxygen – it has nowhere else to go.
Recall the findamental idea that electrostatic charges, whether positive or negative, are more stable when they are ‘spread out’ than when they are confined to one atom. Here, a charge is being ‘spread out’ (in other words, delocalized) by resonance, rather than simply by the size of the atom involved.
The delocalization of charge by resonance has a very powerful effect on the reactivity of organic molecules, enough to account for the difference of over 12 pKa units between ethanol and acetic acid. The acetate ion is that much more stable than the ethoxide ion, all due to the effects of resonance delocalization.
The resonance effect also explains why a nitrogen atom is much more basic when it is in an amine, but not significantly basic when it is part of an amide group. Recall that in an amide, there is significant double-bond character to the carbon-nitrogen bond, due to a second resonance contributor in which the nitrogen lone pair is part of a p bond.
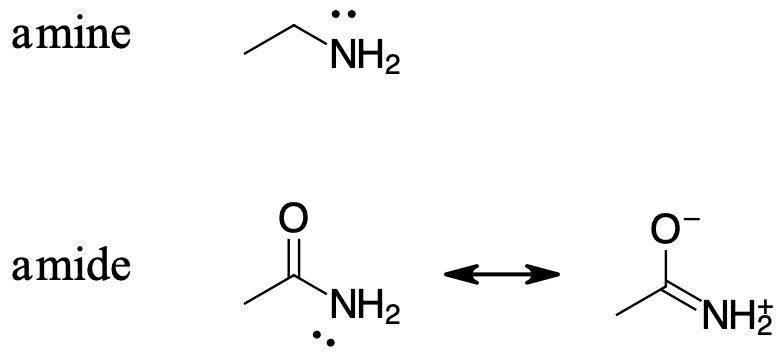
While the electron lone pair of an amine nitrogen is ‘stuck’ in one place, the lone pair on an amide nitrogen is delocalized by resonance. Notice that in this case, we are extending our central statement to say that electron density – in the form of a lone pair – is stabilized by resonance delocalization, even though there is not a negative charge involved. Here’s another way to think about it: the lone pair on an amide nitrogen is not available for bonding with a proton – these two electrons are too ‘comfortable’ being part of the delocalized pi-bonding system. The lone pair on an amine nitrogen, by contrast, is not part of a delocalized pi system, and is very ready to form a bond with any acidic proton that might be nearby.
Why is phenol acidic?
Compounds like alcohols and phenol which contain an -OH group attached to a hydrocarbon are very weak acids. Alcohols are so weakly acidic that, for normal lab purposes, their acidity can be virtually ignored. However, phenol is sufficiently acidic for it to have recognizably acidic properties - even if it is still a very weak acid. A hydrogen ion can break away from the -OH group and transfer to a base. For example, in solution in water:

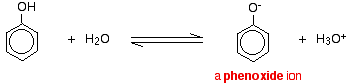
Phenol is a very weak acid and the position of equilibrium lies well to the left. Phenol can lose a hydrogen ion because the phenoxide ion formed is stabilised to some extent. The negative charge on the oxygen atom is delocalised around the ring. The more stable the ion is, the more likely it is to form. One of the lone pairs on the oxygen atom overlaps with the delocalised electrons on the benzene ring.
This overlap leads to a delocalization which extends from the ring out over the oxygen atom. As a result, the negative charge is no longer entirely localized on the oxygen, but is spread out around the whole ion.
Spreading the charge around makes the ion more stable than it would be if all the charge remained on the oxygen. However, oxygen is the most electronegative element in the ion and the delocalized electrons will be drawn towards it. That means that there will still be a lot of charge around the oxygen which will tend to attract the hydrogen ion back again. That is why phenol is only a very weak acid.
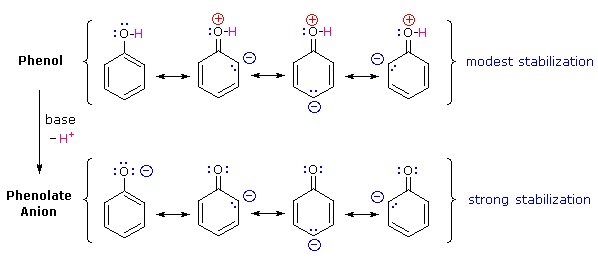
Why is phenol a much stronger acid than cyclohexanol? To answer this question we must evaluate the manner in which an oxygen substituent interacts with the benzene ring. As noted in our earlier treatment of electrophilic aromatic substitution reactions, an oxygen substituent enhances the reactivity of the ring and favors electrophile attack at ortho and para sites. It was proposed that resonance delocalization of an oxygen non-bonded electron pair into the pi-electron system of the aromatic ring was responsible for this substituent effect. A similar set of resonance structures for the phenolate anion conjugate base appears below the phenol structures.

The resonance stabilization in these two cases is very different. An important principle of resonance is that charge separation diminishes the importance of canonical contributors to the resonance hybrid and reduces the overall stabilization. The contributing structures to the phenol hybrid all suffer charge separation, resulting in very modest stabilization of this compound. On the other hand, the phenolate anion is already charged, and the canonical contributors act to disperse the charge, resulting in a substantial stabilization of this species. The conjugate bases of simple alcohols are not stabilized by charge delocalization, so the acidity of these compounds is similar to that of water. An energy diagram showing the effect of resonance on cyclohexanol and phenol acidities is shown on the right. Since the resonance stabilization of the phenolate conjugate base is much greater than the stabilization of phenol itself, the acidity of phenol relative to cyclohexanol is increased. Supporting evidence that the phenolate negative charge is delocalized on the ortho and para carbons of the benzene ring comes from the influence of electron-withdrawing substituents at those sites.
Contributors
- Layne A. Morsch - University of Illinois Springfield