
9.11: Electrons Populate Molecular Orbitals According to the Pauli Exclusion Principle
- Page ID
- 143035
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)The Pauli exclusion principle plays as important a role in the understanding of the electronic structure of molecules as it does in the case of atoms. The end result of the Pauli principle is to limit the amount of electronic charge density that can be placed at any one point in space. For example, the Pauli principle prevents the 1s orbital in an atom from containing more than two electrons. Since the 1s orbital places most of its charge density in regions close to the nucleus, the Pauli principle, by limiting the occupation of the 1s orbital, limits the amount of density close to the nucleus. Any remaining electrons must be placed in orbitals which concentrate their charge density further from the nucleus.
In an earlier discussion we pointed out that the reason the electron doesn't fall onto the nucleus is because it must possess kinetic energy if Heisenberg's uncertainty principle is not to be violated. This is one reason why matter doesn't collapse. The Pauli principle is equally important in this regard. The electron density of the outer electrons in an atom cannot collapse and move closer to the nucleus since it can do so only if the electrons occupy an orbital with a lower n value. If, however, the inner orbital contains two electrons, then the Pauli principle states that the collapse cannot occur. We must be careful in our interpretation of this aspect of the Pauli principle. The density from a 2s orbital has a small but finite probability of being found well within the density of the 1s orbital. Do not interpret the Pauli principle as implying that the density from an occupied orbital has a clearly defined and distinct region in real space all to its own. This is not the case. The operation of the Pauli principle is more subtle than this. In some simple cases, such as the ones we wish to discuss below, the limiting effect of the Pauli principle on the density distribution can, however, be calculated and pictured in a very direct manner.
The Pauli principle demands that when two electrons are placed in the same orbital their spins must be paired. What restriction is placed on the spins of the electrons during the formation of a molecule, when two orbitals, each on a different atom, overlap one another? For example, consider the approach of two hydrogen atoms to form a hydrogen molecule. Consider atom A to have the configuration \(1s^1 \alpha\) and atom B the configuration \(1s^1 \beta\). Even when the atoms approach very close to one another the Pauli principle would be satisfied as the spins of the two electrons are opposed. This is the situation we have tacitly assumed in our previous discussion of the hydrogen molecule. However, what would occur if two hydrogen atoms approached one another and both had the same configuration and spin, say \(1s^1 \alpha\)? When two atoms are relatively close together the electrons become indistinguishable. It is no longer possible to say which electron is associated with which atom as both electrons move in the vicinity of both nuclei. Indeed this is the effect which gives rise to the chemical bond. In so far as we can still regard the region around each atom to be governed by its own atomic orbital, distorted as it may be, two electrons with the same spin will not be able to concentrate their density in the binding region. This region is common to the orbitals on both atoms, and since the electrons possess the same spin they cannot both be there simultaneously. In the region of greatest overlap of the orbitals, the binding region, the presence of one electron will tend to exclude the presence of the other if their spins are parallel. Instead of density accumulating in the binding region as two atoms approach, electron density is removed from this region and placed in the antibonding region behind each nucleus where the overlap of the orbitals is much smaller. Thus the approach of two hydrogen atoms with parallel spins does not result in the formation of a stable molecule. This repulsive state of the hydrogen molecule, in which both electrons have the same spin and atomic orbital quantum numbers, can be detected spectroscopically.
We can now give the general requirements for the formation of a chemical bond. Electron density must be accumulated in the region between the nuclei to an extent greater than that obtained by allowing the original atomic density distributions to overlap. In general, the increase in charge density necessary to balance the nuclear force of repulsion requires the presence of two electrons.
We are now in a position to build up and determine the electronic configurations of the homonuclear diatomic molecules by adding electrons two at a time to the molecular orbitals with the spins of the electrons paired, always filling the orbitals of lowest energy first. We shall, at the same time, discuss the effectiveness of each orbital in binding the nuclei and make qualitative predictions regarding the stability of each molecular configuration.
The Pauli Exclusion Principle in Hydrogen Dimer
The two electrons in the hydrogen molecule may both be accommodated in the 1sg orbital if their spins are paired and the molecular orbital configuration for H2 is 1sg2. Since the 1sg orbital is the only occupied orbital in the ground state of H2, the density distribution shown previously in Figure 9.9.2 for H2 is also the density distribution for the 1sg orbital when occupied by two electrons. The remarks made previously regarding the binding of the nuclei in H2 by the molecular charge distribution apply directly to the properties of the 1sg charge density.
The Pauli Exclusion Principle in Helium Dimer
The electronic configuration of He2 is 1sg2 1su2. A su orbital, unlike a sg orbital, possesses a node in the plane midway between the nuclei and perpendicular to the bond axis. The 1su orbital and all su orbitals in general, because of this nodal property, cannot concentrate charge density in the binding region. It is instead concentrated in the antibinding region behind each nucleus (Figure 9.9.3 ).
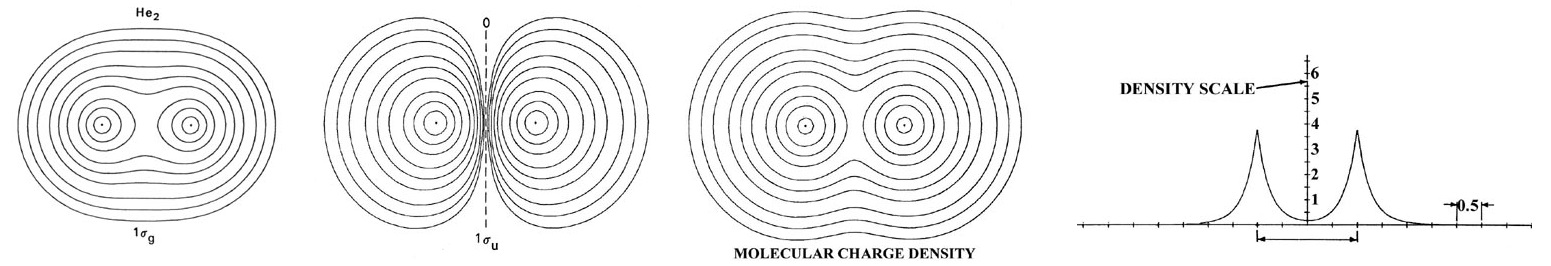
The su orbitals are therefore classified as antibonding. It is evident from the form of density distribution for the 1su orbital that the charge density in this orbital pulls the nuclei apart rather than drawing them together. Generally, the occupation of an equal number of sg and su orbitals results in an unstable molecule. The attractive force exerted on the nuclei by the charge density in the sg orbitals is not sufficient to balance both the nuclear force of repulsion and the antibinding force exerted by the density in the su orbitals. Thus molecular orbital theory ascribes the instability of He2 to the equal occupation of bonding and antibonding orbitals. Notice that the Pauli exclusion principle is still the basic cause of the instability. If it were not for the Pauli principle, all four electrons could occupy a sg-type orbital and concentrate their charge density in the region of low potential energy between the nuclei. It is the Pauli principle, and not a question of energetics, which forces the occupation of the 1su antibonding orbital.
The total molecular charge distribution is obtained by summing the individual molecular orbital densities for single or double occupation numbers as determined by the electronic configuration of the molecule. Thus the total charge distribution for He2 (Figure 9.9.3 ) is given by the sum of the 1sg and 1su orbital densities for double occupation of both orbitals. The adverse effect which the nodal property of the 1su orbital has on the stability of He2 is very evident in the total charge distribution. Very little charge density is accumulated in the central portion of the binding region. The value of the charge density at the mid-point of the bond in He2 is only 0.164 au compared to a value of 0.268 au for H2.
We should reconsider in the light of molecular orbital theory the stability of \(He_2^+\) and the instability of the hydrogen molecule with parallel spins. He2+ will have the configuration 1sg2 1su1. Since the 1su orbital is only singly occupied in \(He_2^+\), less charge density is accumulated in the antibinding regions than is accumulated in these same regions in the neutral molecule. Thus the binding forces of the doubly-occupied 1sg density predominate and \(He_2^+\) is stable. The electron configuration of (triplet) \(H_2\) is \(1s_g^1(\alpha)1s_u^1(\alpha)\) when the electronic spins are parallel. The electrons must occupy separate orbitals because of the Pauli exclusion principle. With equal occupation of bonding and antibonding orbitals, the triplet \(H_2\) species is predicted to be unstable.