
18.7: ΔG° and K as Functions of Temperature
- Page ID
- 46239
- To know the relationship between free energy and the equilibrium constant.
As was previously demonstrated, the spontaneity of a process may depend upon the temperature of the system. Phase transitions, for example, will proceed spontaneously in one direction or the other depending upon the temperature of the substance in question. Likewise, some chemical reactions can also exhibit temperature dependent spontaneities. To illustrate this concept, the equation relating free energy change to the enthalpy and entropy changes for the process is considered:
\[ ΔG=ΔH−TΔS \]
The spontaneity of a process, as reflected in the arithmetic sign of its free energy change, is then determined by the signs of the enthalpy and entropy changes and, in some cases, the absolute temperature. Since T is the absolute (kelvin) temperature, it can only have positive values. Four possibilities therefore exist with regard to the signs of the enthalpy and entropy changes:
- Both ΔH and ΔS are positive. This condition describes an endothermic process that involves an increase in system entropy. In this case, ΔG will be negative if the magnitude of the TΔS term is greater than ΔH. If the TΔS term is less than ΔH, the free energy change will be positive. Such a process is spontaneous at high temperatures and nonspontaneous at low temperatures.
- Both ΔH and ΔS are negative. This condition describes an exothermic process that involves a decrease in system entropy. In this case, ΔG will be negative if the magnitude of the TΔS term is less than ΔH. If the TΔS term’s magnitude is greater than ΔH, the free energy change will be positive. Such a process is spontaneous at low temperatures and nonspontaneous at high temperatures.
- ΔH is positive and ΔS is negative. This condition describes an endothermic process that involves a decrease in system entropy. In this case, ΔG will be positive regardless of the temperature. Such a process is nonspontaneous at all temperatures.
- ΔH is negative and ΔS is positive. This condition describes an exothermic process that involves an increase in system entropy. In this case, ΔG will be negative regardless of the temperature. Such a process is spontaneous at all temperatures.
These four scenarios are summarized in Figure \(\PageIndex{1}\).

The incomplete combustion of carbon is described by the following equation:
\[\ce{2C}(s)+\ce{O2}(g)⟶\ce{2CO}(g) \nonumber\]
How does the spontaneity of this process depend upon temperature?
Solution
Combustion processes are exothermic (ΔH < 0). This particular reaction involves an increase in entropy due to the accompanying increase in the amount of gaseous species (net gain of one mole of gas, ΔS > 0). The reaction is therefore spontaneous (ΔG < 0) at all temperatures.
Popular chemical hand warmers generate heat by the air-oxidation of iron:
How does the spontaneity of this process depend upon temperature?
Answer
ΔH and ΔS are negative; the reaction is spontaneous at low temperatures.
When considering the conclusions drawn regarding the temperature dependence of spontaneity, it is important to keep in mind what the terms “high” and “low” mean. Since these terms are adjectives, the temperatures in question are deemed high or low relative to some reference temperature. A process that is nonspontaneous at one temperature but spontaneous at another will necessarily undergo a change in “spontaneity” (as reflected by its ΔG) as temperature varies. This is clearly illustrated by a graphical presentation of the free energy change equation, in which ΔG is plotted on the y axis versus T on the x axis:
\[ΔG=ΔH−TΔS\]
\[y=b+mx\]
Such a plot is shown in Figure \(\PageIndex{2}\). A process whose enthalpy and entropy changes are of the same arithmetic sign will exhibit a temperature-dependent spontaneity as depicted by the two yellow lines in the plot. Each line crosses from one spontaneity domain (positive or negative ΔG) to the other at a temperature that is characteristic of the process in question. This temperature is represented by the x-intercept of the line, that is, the value of T for which ΔG is zero:
\[ΔG=0=ΔH−TΔS\]
\[T=\dfrac{ΔH}{ΔS}\]
And so, saying a process is spontaneous at “high” or “low” temperatures means the temperature is above or below, respectively, that temperature at which ΔG for the process is zero. As noted earlier, this condition describes a system at equilibrium.

As defined in the chapter on liquids and solids, the boiling point of a liquid is the temperature at which its solid and liquid phases are in equilibrium (that is, when vaporization and condensation occur at equal rates). Use the information in Appendix G to estimate the boiling point of water.
Solution
The process of interest is the following phase change:
When this process is at equilibrium, ΔG = 0, so the following is true:
Using the standard thermodynamic data from Appendix G,
ΔH°&=ΔH^\circ_\ce{f}(\ce{H2O}(g))−ΔH^\circ_\ce{f}(\ce{H2O}(l)) \nonumber\\
&=\mathrm{−241.82\: kJ/mol−(−285.83\: kJ/mol)=44.01\: kJ/mol} \nonumber
\end{align}\]
ΔS°&=ΔS^\circ_{298}(\ce{H2O}(g))−ΔS^\circ_{298}(\ce{H2O}(l)) \nonumber\\
&=\mathrm{188.8\: J/K⋅mol−70.0\: J/K⋅mol=118.8\: J/K⋅mol} \nonumber
\end{align}\]
The accepted value for water’s normal boiling point is 373.2 K (100.0 °C), and so this calculation is in reasonable agreement. Note that the values for enthalpy and entropy changes data used were derived from standard data at 298 K (Appendix G). If desired, you could obtain more accurate results by using enthalpy and entropy changes determined at (or at least closer to) the actual boiling point.
Use the information in Appendix G to estimate the boiling point of CS2.
Answer
313 K (accepted value 319 K)
Temperature Dependence of the Equilibrium Constant
The fact that ΔG° and K are related provides us with another explanation of why equilibrium constants are temperature dependent. This relationship can be expressed as follows:
\[\ln K=-\dfrac{\Delta H^\circ}{RT}+\dfrac{\Delta S^\circ}{R} \label{18.40}\]
Assuming ΔH° and ΔS° are temperature independent, for an exothermic reaction (ΔH° < 0), the magnitude of K decreases with increasing temperature, whereas for an endothermic reaction (ΔH° > 0), the magnitude of K increases with increasing temperature. The quantitative relationship expressed in Equation \(\ref{18.40}\) agrees with the qualitative predictions made by applying Le Chatelier’s principle. Because heat is produced in an exothermic reaction, adding heat (by increasing the temperature) will shift the equilibrium to the left, favoring the reactants and decreasing the magnitude of K. Conversely, because heat is consumed in an endothermic reaction, adding heat will shift the equilibrium to the right, favoring the products and increasing the magnitude of K. Equation \(\ref{18.40}\) also shows that the magnitude of ΔH° dictates how rapidly K changes as a function of temperature. In contrast, the magnitude and sign of ΔS° affect the magnitude of K but not its temperature dependence.
If we know the value of K at a given temperature and the value of ΔH° for a reaction, we can estimate the value of K at any other temperature, even in the absence of information on ΔS°. Suppose, for example, that K1 and K2 are the equilibrium constants for a reaction at temperatures T1 and T2, respectively. Applying Equation \(\ref{18.40}\) gives the following relationship at each temperature:
\\ \ln K_2 &=\dfrac{-\Delta H^\circ}{RT_2}+\dfrac{\Delta S^\circ}{R}\end{align}\]
Subtracting \(\ln K_1\) from \(\ln K_2\),
\[\ln K_2-\ln K_1=\ln\dfrac{K_2}{K_1}=\dfrac{\Delta H^\circ}{R}\left(\dfrac{1}{T_1}-\dfrac{1}{T_2}\right) \label{18.41}\]
Thus calculating ΔH° from tabulated enthalpies of formation and measuring the equilibrium constant at one temperature (K1) allow us to calculate the value of the equilibrium constant at any other temperature (K2), assuming that ΔH° and ΔS° are independent of temperature. The linear relation between \(\ln K \)and the standard enthalpies and entropies in Equation \(\ref{18.41}\) is known as the van’t Hoff equation. It shows that a plot of \(\ln K\) vs. \(1/T\) should be a line with slope \(-\Delta_r{H^o}/R\) and intercept \(\Delta_r{S^o}/R\).
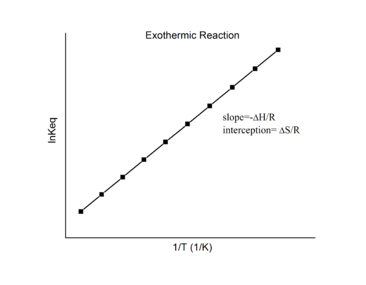
Hence, these thermodynamic enthalpy and entropy changes for a reversible reaction can be determined from plotting \(\ln K\) vs. \(1/T\) data without the aid of calorimetry. Of course, the main assumption here is that \(\Delta_r{H^o}\) and \(\Delta_r{S^o}\) are only very weakly dependent on \(T\), which is usually valid over a narrow temperature range.
The equilibrium constant for the formation of NH3 from H2 and N2 at 25°C is Kp = 5.4 × 105. What is Kp at 500°C? (Use the data from Example 10.)
Given: balanced chemical equation, ΔH°, initial and final T, and Kp at 25°C
Asked for: Kp at 500°C
Strategy:
Convert the initial and final temperatures to kelvins. Then substitute appropriate values into Equation \(\ref{18.41}\) to obtain K2, the equilibrium constant at the final temperature.
Solution:
The value of ΔH° for the reaction obtained using Hess’s law is −91.8 kJ/mol of N2. If we set T1 = 25°C = 298.K and T2 = 500°C = 773 K, then from Equation \(\ref{18.41}\) we obtain the following:
\\&=\dfrac{(-\textrm{91.8 kJ})(\textrm{1000 J/kJ})}{\textrm{8.314 J/K}}\left(\dfrac{1}{\textrm{298 K}}-\dfrac{1}{\textrm{773 K}}\right)=-22.8
\\ \dfrac{K_2}{K_1}&=1.3\times10^{-10}
\\ K_2&=(5.4\times10^5)(1.3\times10^{-10})=7.0\times10^{-5}\end{align}\)
Thus at 500°C, the equilibrium strongly favors the reactants over the products.
In the exercise in Example \(\PageIndex{3}\), you calculated Kp = 2.2 × 1012 for the reaction of NO with O2 to give NO2 at 25°C. Use the ΔH∘f values in the exercise in Example 10 to calculate Kp for this reaction at 1000°C.
Answer: 5.6 × 10−4
The Van't Hoff Equation: https://youtu.be/4vk6idAXp_A
Summary
For a reversible process that does not involve external work, we can express the change in free energy in terms of volume, pressure, entropy, and temperature. If we assume ideal gas behavior, the ideal gas law allows us to express ΔG in terms of the partial pressures of the reactants and products, which gives us a relationship between ΔG and Kp, the equilibrium constant of a reaction involving gases, or K, the equilibrium constant expressed in terms of concentrations. If ΔG° < 0, then K or Kp > 1, and products are favored over reactants. If ΔG° > 0, then K or Kp < 1, and reactants are favored over products. If ΔG° = 0, then K or Kp = 1, and the system is at equilibrium. We can use the measured equilibrium constant K at one temperature and ΔH° to estimate the equilibrium constant for a reaction at any other temperature.
Contributors and Attributions
- Anonymous