
18.6: Gibbs Energy Change and Equilibrium
- Page ID
- 46238
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)- To know the relationship between free energy and the equilibrium constant.
- The sign of the standard free energy change ΔG° of a chemical reaction determines whether the reaction will tend to proceed in the forward or reverse direction.
- Similarly, the relative signs of ΔG° and ΔS° determine whether the spontaniety of a chemical reaction will be affected by the temperature, and if so, in what way.
ΔG is meaningful only for changes in which the temperature and pressure remain constant. These are the conditions under which most reactions are carried out in the laboratory; the system is usually open to the atmosphere (constant pressure) and we begin and end the process at room temperature (after any heat we have added or which is liberated by the reaction has dissipated.) The importance of the Gibbs function can hardly be over-stated: it serves as the single master variable that determines whether a given chemical change is thermodynamically possible. Thus if the free energy of the reactants is greater than that of the products, the entropy of the world will increase when the reaction takes place as written, and so the reaction will tend to take place spontaneously. Conversely, if the free energy of the products exceeds that of the reactants, then the reaction will not take place in the direction written, but it will tend to proceed in the reverse direction.
Temperature Dependence to ΔG
In a spontaneous change, Gibbs energy always decreases and never increases. This of course reflects the fact that the entropy of the world behaves in the exact opposite way (owing to the negative sign in the TΔS term).
\[H_2O_{(l)} \rightarrow H_2O_{(s)} \label{23.5.6}\]
water below its freezing point undergoes a decrease in its entropy, but the heat released into the surroundings more than compensates for this, so the entropy of the world increases, the free energy of the H2O diminishes, and the process proceeds spontaneously.
In a spontaneous change, Gibbs energy always decreases and never increases.
An important consequence of the one-way downward path of the free energy is that once it reaches its minimum possible value, all net change comes to a halt. This, of course, represents the state of chemical equilibrium. These relations are nicely summarized as follows:
- ΔG < 0: reaction can spontaneously proceed to the right: \[A \rightarrow B\]
- ΔG > 0: reaction can spontaneously proceed to the left: \[A \leftarrow B\]
- ΔG = 0: the reaction is at equilibrium; the quantities of [A] and [B] will not change
Recall the condition for spontaneous change
\[ΔG = ΔH – TΔS < 0 \label{Master}\]
it is apparent that the temperature dependence of ΔG depends almost entirely on the entropy change associated with the process. (We say "almost" because the values of ΔH and ΔS are themselves slightly temperature dependent; both gradually increase with temperature). In particular, notice that in the above equation the sign of the entropy change determines whether the reaction becomes more or less spontaneous as the temperature is raised. For any given reaction, the sign of ΔH can also be positive or negative. This means that there are four possibilities for the influence that temperature can have on the spontaneity of a process.
The following cases generalizes these relations for the four sign-combinations of ΔH and ΔS. (Note that use of the standard ΔH° and ΔS° values in the example reactions is not strictly correct here, and can yield misleading results when used generally.)
> 0
Under these conditions, both the ΔH and TΔS terms will be negative, so ΔG will be negative regardless of the temperature. An exothermic reaction whose entropy increases will be spontaneous at all temperatures.

Example Reaction
\[C_{(graphite)} + O_{2(g)} \rightleftharpoons CO_{2(g)}\]
- ΔH° = –393 kJ
- ΔS° = +2.9 J K–1
- ΔG° = –394 kJ at 298 K
The positive entropy change is due mainly to the greater mass of CO2 molecules compared to those of O2.
< 0
If the reaction is sufficiently exothermic it can force ΔG negative only at temperatures below which |TΔS| < |ΔH|. This means that there is a temperature T = ΔH / ΔS at which the reaction is at equilibrium; the reaction will only proceed spontaneously below this temperature. The freezing of a liquid or the condensation of a gas are the most common examples of this condition.
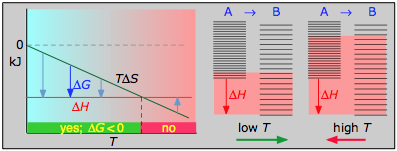
Example reaction:
\[3 H_2 + N_2 \rightleftharpoons 2 NH_{3(g)}\]
- ΔH° = –46.2 kJ
- ΔS° = –389 J K–1
- ΔG° = –16.4 kJ at 298 K
The decrease in moles of gas in the Haber ammonia synthesis drives the entropy change negative, making the reaction spontaneous only at low temperatures. Thus higher T, which speeds up the reaction, also reduces its extent.
> 0
This is the reverse of the previous case; the entropy increase must overcome the handicap of an endothermic process so that TΔS > ΔH. Since the effect of the temperature is to "magnify" the influence of a positive ΔS, the process will be spontaneous at temperatures above T = ΔH / ΔS. (Think of melting and boiling.)
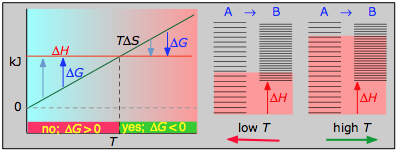
Example reaction:
\[N_2O_{4(g)} \rightleftharpoons 2 NO_{2(g)}\]
- ΔH° = 55.3 kJ
- ΔS° = +176 J K–1
- ΔG° = +2.8 kJ at 298 K
Dissociation reactions are typically endothermic with positive entropy change, and are therefore spontaneous at high temperatures.Ultimately, all molecules decompose to their atoms at sufficiently high temperatures.
< 0
With both ΔH and ΔS working against it, this kind of process will not proceed spontaneously at any temperature. Substance A always has a greater number of accessible energy states, and is therefore always the preferred form.
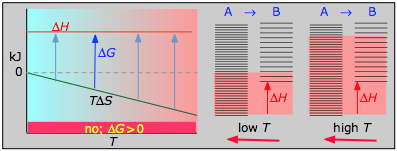
Example reaction:
\[½ N_2 + O_2 \rightleftharpoons NO_{2(g)}\]
- ΔH° = 33.2 kJ
- ΔS° = –249 J K–1
- ΔG° = +51.3 kJ at 298 K
This reaction is not spontaneous at any temperature, meaning that its reverse is always spontaneous. But because the reverse reaction is kinetically inhibited, NO2 can exist indefinitely at ordinary temperatures even though it is thermodynamically unstable.
The above cases and associated plots are the important ones; do not try to memorize them, but make sure you understand and can explain or reproduce them for a given set of ΔH and ΔS.
- Their most important differentiating features are the position of the ΔH line (above or below the is TΔS line), and the slope of the latter, which of course depends on the sign of ΔS.
- The reaction A → B will occur spontaneously only when ΔG is negative (blue arrows pointing down.)
- Owing to the slight temperature dependence of ΔS, the TΔS plots are not quite straight lines as shown here. Similarly, the lines representing ΔH are even more curved.
The other two plots on each diagram are only for the chemistry-committed.
- Each pair of energy-level diagrams depicts the relative spacing of the microscopic energy levels in the reactants and products as reflected by the value of ΔS°. (The greater the entropy, the more closely-spaced are the quantized microstates.)
- The red shading indicates the range of energy levels that are accessible to the system at each temperature. The spontaneous direction of the reaction will always be in the direction in which the red shading overlaps the greater number of energy levels, resulting in the maximum dispersal of thermal energy.
- Note that the vertical offsets correspond to ΔH° for the reaction.
- Never forget that it is the ability of thermal energy to spread into as many of these states as possible that determines the tendency of the process to take place. None of this is to scale, of course!
Liquid-Vapor Equilibrium
To further understand how the various components of ΔG dictate whether a process occurs spontaneously, we now look at a simple and familiar physical change: the conversion of liquid water to water vapor. If this process is carried out at 1 atm and the normal boiling point of 100.00°C (373.15 K), we can calculate ΔG from the experimentally measured value of ΔHvap (40.657 kJ/mol). For vaporizing 1 mol of water, \(ΔH = 40,657; J\), so the process is highly endothermic. From the definition of ΔS, we know that for 1 mol of water,
Hence there is an increase in the disorder of the system. At the normal boiling point of water,
\[\Delta G_{100^\circ\textrm C}=\Delta H_{100^\circ\textrm C}-T\Delta S_{100^\circ\textrm C}=\textrm{40,657 J}-[(\textrm{373.15 K})(\textrm{108.96 J/K})]=\textrm{0 J}\]
The energy required for vaporization offsets the increase in disorder of the system. Thus ΔG = 0, and the liquid and vapor are in equilibrium, as is true of any liquid at its boiling point under standard conditions.
Now suppose we were to superheat 1 mol of liquid water to 110°C. The value of ΔG for the vaporization of 1 mol of water at 110°C, assuming that ΔH and ΔS do not change significantly with temperature, becomes
\[\Delta G_{110^\circ\textrm C}=\Delta H-T\Delta S=\textrm{40,657 J}-[(\textrm{383.15 K})(\textrm{108.96 J/K})]=-\textrm{1091 J}\]
At 110°C, ΔG < 0, and vaporization is predicted to occur spontaneously and irreversibly.
We can also calculate ΔG for the vaporization of 1 mol of water at a temperature below its normal boiling point—for example, 90°C—making the same assumptions:
\[\Delta G_{90^\circ\textrm C}=\Delta H-T\Delta S=\textrm{40,657 J}-[(\textrm{363.15 K})(\textrm{108.96 J/K})]=\textrm{1088 J}\]
At 90°C, ΔG > 0, and water does not spontaneously convert to water vapor. When using all the digits in the calculator display in carrying out our calculations, ΔG110°C = 1090 J = −ΔG90°C, as we would predict.
We can also calculate the temperature at which liquid water is in equilibrium with water vapor. Inserting the values of ΔH and ΔS into the definition of ΔG (Equation \(\ref{Master}\)), setting ΔG = 0, and solving for T,
Thus ΔG = 0 at T = 373.15 K and 1 atm, which indicates that liquid water and water vapor are in equilibrium; this temperature is called the normal boiling point of water. At temperatures greater than 373.15 K, ΔG is negative, and water evaporates spontaneously and irreversibly. Below 373.15 K, ΔG is positive, and water does not evaporate spontaneously. Instead, water vapor at a temperature less than 373.15 K and 1 atm will spontaneously and irreversibly condense to liquid water. Figure \(\PageIndex{1}\) shows how the ΔH and TΔS terms vary with temperature for the vaporization of water. When the two lines cross, ΔG = 0, and ΔH = TΔS.

A similar situation arises in the conversion of liquid egg white to a solid when an egg is boiled. The major component of egg white is a protein called albumin, which is held in a compact, ordered structure by a large number of hydrogen bonds. Breaking them requires an input of energy (ΔH > 0), which converts the albumin to a highly disordered structure in which the molecules aggregate as a disorganized solid (ΔS > 0). At temperatures greater than 373 K, the TΔS term dominates, and ΔG < 0, so the conversion of a raw egg to a hard-boiled egg is an irreversible and spontaneous process above 373 K.
Free Energy and the Equilibrium Constant
ΔG is key in determining whether or not a reaction will take place in a given direction. It turns out, however, that it is almost never necessary to explicitly evaluate ΔG. It is far more convenient to work with the equilibrium constant of a reaction, within which ΔG is "hidden". This is just as well, because for most reactions (those that take place in solutions or gas mixtures) the value of ΔG depends on the proportions of the various reaction components in the mixture; it is not a simple sum of the "products minus reactants" type, as is the case with ΔH.
Because ΔH° and ΔS° determine the magnitude of ΔG° and because K is a measure of the ratio of the concentrations of products to the concentrations of reactants, we should be able to express K in terms of ΔG° and vice versa. ΔG is equal to the maximum amount of work a system can perform on its surroundings while undergoing a spontaneous change. For a reversible process that does not involve external work, we can express the change in free energy in terms of volume, pressure, entropy, and temperature, thereby eliminating ΔH from the equation for ΔG. The general relationship can be shown as follow (derivation not shown):
\[ \Delta G = V \Delta P − S \Delta T \label{18.29}\]
If a reaction is carried out at constant temperature (ΔT = 0), then Equation \(\ref{18.29}\) simplifies to
\[\Delta{G} = V\Delta{P} \label{18.30}\]
Under normal conditions, the pressure dependence of free energy is not important for solids and liquids because of their small molar volumes. For reactions that involve gases, however, the effect of pressure on free energy is very important.
Assuming ideal gas behavior, we can replace the \(V\) in Equation \(\ref{18.30}\) by nRT/P (where n is the number of moles of gas and R is the ideal gas constant) and express \(\Delta{G}\) in terms of the initial and final pressures (\(P_i\) and \(P_f\), respectively):
\[\Delta G=\left(\dfrac{nRT}{P}\right)\Delta P=nRT\dfrac{\Delta P}{P}=nRT\ln\left(\dfrac{P_\textrm f}{P_\textrm i}\right) \label{18.31}\]
If the initial state is the standard state with Pi = 1 atm, then the change in free energy of a substance when going from the standard state to any other state with a pressure P can be written as follows:
\[G − G^° = nRT\ln{P}\]
This can be rearranged as follows:
\[G = G^° + nRT\ln {P} \label{18.32}\]
As you will soon discover, Equation \(\ref{18.32}\) allows us to relate ΔG° and Kp. Any relationship that is true for \(K_p\) must also be true for \(K\) because \(K_p\) and \(K\) are simply different ways of expressing the equilibrium constant using different units.
Let’s consider the following hypothetical reaction, in which all the reactants and the products are ideal gases and the lowercase letters correspond to the stoichiometric coefficients for the various species:
\[aA+bB \rightleftharpoons cC+dD \label{18.33}\]
Because the free-energy change for a reaction is the difference between the sum of the free energies of the products and the reactants, we can write the following expression for ΔG:
\[\delta{G}=\sum_m G_{products}−\sum_n G_{reactants}=(cG_C+dG_D)−(aG_A+bG_B) \label{18.34}\]
Substituting Equation \(\ref{18.32}\) for each term into Equation \(\ref{18.34}\),
\[ΔG=[(cG^o_C+cRT \ln P_C)+(dG^o_D+dRT\ln P_D)]−[(aG^o_A+aRT\ln P_A)+(bG^o_B+bRT\ln P_B)]\]
Combining terms gives the following relationship between ΔG and the reaction quotient Q:
\[\Delta G=\Delta G^\circ+RT \ln\left(\dfrac{P^c_\textrm CP^d_\textrm D}{P^a_\textrm AP^b_\textrm B}\right)=\Delta G^\circ+RT\ln Q \label{18.35}\]
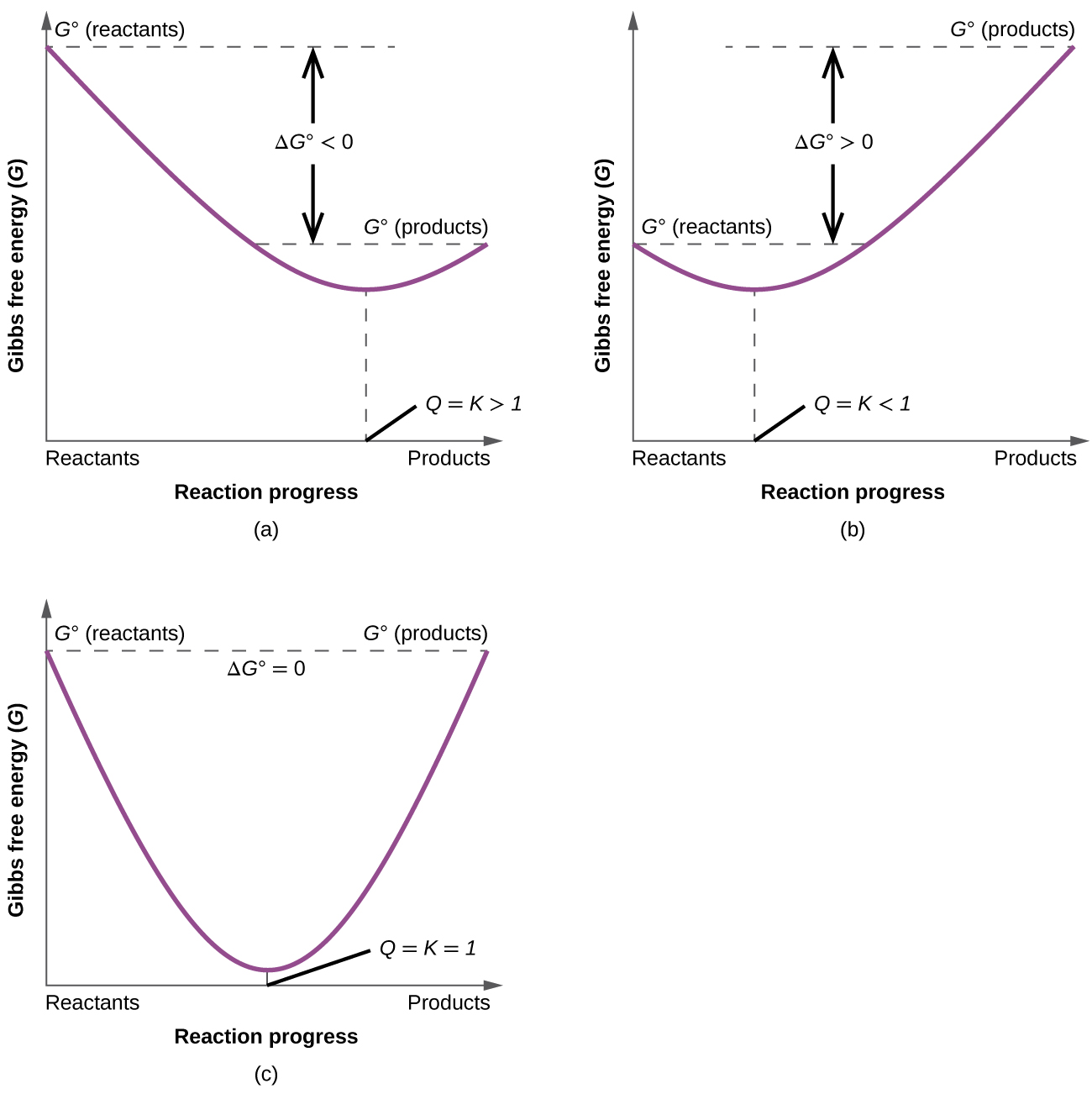
where ΔG° indicates that all reactants and products are in their standard states. For gases at equilibrium (\(Q = K_p\),), and as you’ve learned in this chapter, ΔG = 0 for a system at equilibrium. Therefore, we can describe the relationship between ΔG° and Kp for gases as follows:
\[ 0 = ΔG° + RT\ln K_p \label{18.36a}\]
\[ \color{red} ΔG°= −RT\ln K_p \label{18.36b}\]
If the products and reactants are in their standard states and ΔG° < 0, then Kp > 1, and products are favored over reactants. Conversely, if ΔG° > 0, then Kp < 1, and reactants are favored over products. If ΔG° = 0, then \(K_p = 1\), and neither reactants nor products are favored: the system is at equilibrium.
For a spontaneous process under standard conditions, \(K_{eq}\) and \(K_p\) are greater than 1.
We previosuly calculated that ΔG° = −32.7 kJ/mol of N2 for the reaction
\[N_{2(g)}+3H_{2(g)} \rightleftharpoons 2NH_{3(g)} \nonumber\]
This calculation was for the reaction under standard conditions—that is, with all gases present at a partial pressure of 1 atm and a temperature of 25°C. Calculate ΔG for the same reaction under the following nonstandard conditions:
- \(P_{\textrm N_2}\) = 2.00 atm,
- \(P_{\textrm H_2}\) = 7.00 atm,
- \(P_{\textrm{NH}_3}\) = 0.021 atm,
- and T = 100°C.
Does the reaction favor products or reactants?
Given: balanced chemical equation, partial pressure of each species, temperature, and ΔG°
Asked for: whether products or reactants are favored
Strategy:
- Using the values given and Equation \(\ref{18.35}\), calculate Q.
- Substitute the values of ΔG° and Q into Equation \(\ref{18.35}\) to obtain ΔG for the reaction under nonstandard conditions.
Solution:
A The relationship between ΔG° and ΔG under nonstandard conditions is given in Equation \(\ref{18.35}\). Substituting the partial pressures given, we can calculate Q:
\[Q=\dfrac{P^2_{\textrm{NH}_3}}{P_{\textrm N_2}P^3_{\textrm H_2}}=\dfrac{(0.021)^2}{(2.00)(7.00)^3}=6.4\times10^{-7} \nonumber\]
B Substituting the values of ΔG° and Q into Equation \(\ref{18.35}\),
\(=-77\textrm{ kJ/mol of N}_2\)
Because ΔG < 0 and Q < 1.0, the reaction is spontaneous to the right as written, so products are favored over reactants.
Calculate ΔG for the reaction of nitric oxide with oxygen to give nitrogen dioxide under these conditions: T = 50°C, PNO = 0.0100 atm, \(P_{\mathrm{O_2}}\) = 0.200 atm, and \(P_{\mathrm{NO_2}}\) = 1.00 × 10−4 atm. The value of ΔG° for this reaction is −72.5 kJ/mol of O2. Are products or reactants favored?
Answer: −92.9 kJ/mol of O2; the reaction is spontaneous to the right as written, so products are favored.
Calculate Kp for the reaction of H2 with N2 to give NH3 at 25°C. As calculated in Example 10, ΔG° for this reaction is −32.7 kJ/mol of N2.
Given: balanced chemical equation from Example 10, ΔG°, and temperature
Asked for: Kp
Strategy:
Substitute values for ΔG° and T (in kelvins) into Equation \(\ref{18.36}\) to calculate Kp, the equilibrium constant for the formation of ammonia.
Solution
In Example 10, we used tabulated values of ΔG∘f to calculate ΔG° for this reaction (−32.7 kJ/mol of N2). For equilibrium conditions, rearranging Equation \(\ref{18.36b}\),
\\ \dfrac{-\Delta G^\circ}{RT} &=\ln K_\textrm p \nonumber \end{align}\)
Inserting the value of ΔG° and the temperature (25°C = 298 K) into this equation,
\nonumber \\ K_\textrm p &=5.4\times10^5 \nonumber\end{align}\)
Thus the equilibrium constant for the formation of ammonia at room temperature is favorable. However, the rate at which the reaction occurs at room temperature is too slow to be useful.
Calculate Kp for the reaction of NO with O2 to give NO2 at 25°C. As calculated in the exercise in Example 10, ΔG° for this reaction is −70.5 kJ/mol of O2.
Answer: 2.2 × 1012
Although Kp is defined in terms of the partial pressures of the reactants and the products, the equilibrium constant K is defined in terms of the concentrations of the reactants and the products. We described the relationship between the numerical magnitude of Kp and K in Chapter 15 and showed that they are related:
\[K_p = K(RT)^{Δn} \label{18.37}\]
where Δn is the number of moles of gaseous product minus the number of moles of gaseous reactant. For reactions that involve only solutions, liquids, and solids, Δn = 0, so Kp = K. For all reactions that do not involve a change in the number of moles of gas present, the relationship in Equation \(\ref{18.36b}\) can be written in a more general form:
\[ΔG° = −RT \ln K \label{18.38}\]
Only when a reaction results in a net production or consumption of gases is it necessary to correct Equation \(\ref{18.38}\) for the difference between Kp and K. Although we typically use concentrations or pressures in our equilibrium calculations, recall that equilibrium constants are generally expressed as unitless numbers because of the use of activities or fugacities in precise thermodynamic work. Systems that contain gases at high pressures or concentrated solutions that deviate substantially from ideal behavior require the use of fugacities or activities, respectively.
Combining Equations \(\ref{18.38}\) with \(ΔG^o = ΔH^o − TΔS^o\) provides insight into how the components of ΔG° influence the magnitude of the equilibrium constant:
\[ΔG° = ΔH° − TΔS° = −RT \ln K \label{18.39}\]
Equation \(\ref{18.39}\) is quite powerful and connected the nature of the system under equilibrium \(K\) to the condition of the system under standard conditions \(\Delta G^o\).; that is quite powerful. Notice that \(K\) becomes larger as ΔS° becomes more positive, indicating that the magnitude of the equilibrium constant is directly influenced by the tendency of a system to move toward maximum disorder. Moreover, K increases as ΔH° decreases. Thus the magnitude of the equilibrium constant is also directly influenced by the tendency of a system to seek the lowest energy state possible.
The magnitude of the equilibrium constant is directly influenced by the tendency of a system to move toward maximum entropy and seek the lowest energy state possible.
To further illustrate the relation between these two essential thermodynamic concepts, consider the observation that reactions spontaneously proceed in a direction that ultimately establishes equilibrium. As may be shown by plotting the free energy change versus the extent of the reaction (for example, as reflected in the value of Q), equilibrium is established when the system’s free energy is minimized (Figure \(\PageIndex{2}\)). If a system is present with reactants and products present in nonequilibrium amounts (Q ≠ K), the reaction will proceed spontaneously in the direction necessary to establish equilibrium.
Relating Grxn and Kp: https://youtu.be/T-OYNTYN__4
ΔG° and ΔG: Predicting the Direction of Chemical Change
We have seen that there is no way to measure absolute enthalpies, although we can measure changes in enthalpy (ΔH) during a chemical reaction. Because enthalpy is one of the components of Gibbs free energy, we are consequently unable to measure absolute free energies; we can measure only changes in free energy. The standard free-energy change (ΔG°) is the change in free energy when one substance or a set of substances in their standard states is converted to one or more other substances, also in their standard states. The standard free-energy change can be calculated from the definition of free energy, if the standard enthalpy and entropy changes are known, using Equation \(\ref{Eq5}\):
\[ΔG° = ΔH° − TΔS° \label{Eq5}\]
If ΔS° and ΔH° for a reaction have the same sign, then the sign of ΔG° depends on the relative magnitudes of the ΔH° and TΔS° terms. It is important to recognize that a positive value of ΔG° for a reaction does not mean that no products will form if the reactants in their standard states are mixed; it means only that at equilibrium the concentrations of the products will be less than the concentrations of the reactants.
A positive ΔG° means that the equilibrium constant is less than 1.
Calculate the standard free-energy change (ΔG°) at 25°C for the reaction
\[H_{2(g)}+O_{2(g)} \rightleftharpoons H_2O_{2(l)} \nonumber\]
At 25°C, the standard enthalpy change (ΔH°) is −187.78 kJ/mol, and the absolute entropies of the products and reactants are:
- S°(H2O2) = 109.6 J/(mol•K),
- S°(O2) = 205.2 J/(mol•K), and
- S°(H2) = 130.7 J/(mol•K).
Is the reaction spontaneous as written?
Given: balanced chemical equation, ΔH° and S° for reactants and products
Asked for: spontaneity of reaction as written
Strategy:
- Calculate ΔS° from the absolute molar entropy values given.
- Use Equation \(\ref{Eq5}\), the calculated value of ΔS°, and other data given to calculate ΔG° for the reaction. Use the value of ΔG° to determine whether the reaction is spontaneous as written.
Solution
A To calculate ΔG° for the reaction, we need to know ΔH°, ΔS°, and T. We are given ΔH°, and we know that T = 298.15 K. We can calculate ΔS° from the absolute molar entropy values provided using the “products minus reactants” rule:
\nonumber \\ &=[\mathrm{1\;mol\;H_2O_2}\times109.6\;\mathrm{J/(mol\cdot K})]
\nonumber \\ &-\left \{ [\textrm{1 mol H}_2\times130.7\;\mathrm{J/(mol\cdot K)}]+[\textrm{1 mol O}_2\times205.2\;\mathrm{J/(mol\cdot K)}] \right \}
\nonumber \\&=-226.3\textrm{ J/K }(\textrm{per mole of }\mathrm{H_2O_2}) \end{align}\)
As we might expect for a reaction in which 2 mol of gas is converted to 1 mol of a much more ordered liquid, ΔS° is very negative for this reaction.
B Substituting the appropriate quantities into Equation \(\ref{Eq5}\),
\[\begin{align}\Delta G^\circ=\Delta H^\circ -T\Delta S^\circ &=-187.78\textrm{ kJ/mol}-(\textrm{298.15 K}) [-226.3\;\mathrm{J/(mol\cdot K)}\times\textrm{1 kJ/1000 J}] \nonumber \\ &=-187.78\textrm{ kJ/mol}+67.47\textrm{ kJ/mol}=-120.31\textrm{ kJ/mol} \nonumber \end{align}\]
The negative value of ΔG° indicates that the reaction is spontaneous as written. Because ΔS° and ΔH° for this reaction have the same sign, the sign of ΔG° depends on the relative magnitudes of the ΔH° and TΔS° terms. In this particular case, the enthalpy term dominates, indicating that the strength of the bonds formed in the product more than compensates for the unfavorable ΔS° term and for the energy needed to break bonds in the reactants.
Calculate the standard free-energy change (ΔG°) at 25°C for the reaction
\[2H_2(g)+N_2(g) \rightleftharpoons N_2H_4(l) \nonumber \]
. At 25°C, the standard enthalpy change (ΔH°) is 50.6 kJ/mol, and the absolute entropies of the products and reactants are S°(N2H4) = 121.2 J/(mol•K), S°(N2) = 191.6 J/(mol•K), and S°(H2) = 130.7 J/(mol•K). Is the reaction spontaneous as written?
Answer:
149.5 kJ/mol; no
Tabulated values of standard free energies of formation allow chemists to calculate the values of ΔG° for a wide variety of chemical reactions rather than having to measure them in the laboratory. The standard free energy of formation (\(ΔG^∘_f\))of a compound is the change in free energy that occurs when 1 mol of a substance in its standard state is formed from the component elements in their standard states. By definition, the standard free energy of formation of an element in its standard state is zero at 298.15 K. One mole of Cl2 gas at 298.15 K, for example, has \(ΔG^∘_f = 0\). The standard free energy of formation of a compound can be calculated from the standard enthalpy of formation (ΔH∘f) and the standard entropy of formation (ΔS∘f) using the definition of free energy:
\[Δ^o_{f} =ΔH^o_{f} −TΔS^o_{f} \label{Eq6}\]
Using standard free energies of formation to calculate the standard free energy of a reaction is analogous to calculating standard enthalpy changes from standard enthalpies of formation using the familiar “products minus reactants” rule:
\[ΔG^o_{rxn}=\sum mΔG^o_{f} (products)− \sum nΔ^o_{f} (reactants) \label{Eq7a}\]
where m and n are the stoichiometric coefficients of each product and reactant in the balanced chemical equation. A very large negative ΔG° indicates a strong tendency for products to form spontaneously from reactants; it does not, however, necessarily indicate that the reaction will occur rapidly. To make this determination, we need to evaluate the kinetics of the reaction.
Calculate ΔG° for the reaction of isooctane with oxygen gas to give carbon dioxide and water (described in Example 7). Use the following data:
- ΔG°f(isooctane) = −353.2 kJ/mol,
- ΔG°f(CO2) = −394.4 kJ/mol, and
- ΔG°f(H2O) = −237.1 kJ/mol. Is the reaction spontaneous as written?
Given: balanced chemical equation and values of ΔG°f for isooctane, CO2, and H2O
Asked for: spontaneity of reaction as written
Strategy:
Use the “products minus reactants” rule to obtain ΔG∘rxn, remembering that ΔG°f for an element in its standard state is zero. From the calculated value, determine whether the reaction is spontaneous as written.
Solution
The balanced chemical equation for the reaction is as follows:
\[\mathrm{C_8H_{18}(l)}+\frac{25}{2}\mathrm{O_2(g)}\rightarrow\mathrm{8CO_2(g)}+\mathrm{9H_2O(l)} \nonumber\]
We are given ΔG∘f values for all the products and reactants except O2(g). Because oxygen gas is an element in its standard state, ΔG∘f (O2) is zero. Using the “products minus reactants” rule,
\nonumber \\ &=[(\textrm{8 mol})(-394.4\textrm{ kJ/mol})+(\textrm{9 mol})(-237.1\textrm{ kJ/mol})]
\nonumber\\&-\left [(\textrm{1 mol})(-353.2\textrm{ kJ/mol})+\left(\dfrac{25}{2}\;\textrm{mol}\right)(0 \textrm{ kJ/mol}) \right ]
\nonumber \\ &=-4935.9\textrm{ kJ }(\textrm{per mol of }\mathrm{C_8H_{18}}) \nonumber \end{align}\)
Because ΔG° is a large negative number, there is a strong tendency for the spontaneous formation of products from reactants (though not necessarily at a rapid rate). Also notice that the magnitude of ΔG° is largely determined by the ΔG∘f of the stable products: water and carbon dioxide.
Calculate ΔG° for the reaction of benzene with hydrogen gas to give cyclohexane using the following data
- ΔG∘f(benzene) = 124.5 kJ/mol
- ΔG∘f (cyclohexane) = 217.3 kJ/mol.
Is the reaction spontaneous as written?
Answer:
- 92.8 kJ; no
Calculated values of ΔG° are extremely useful in predicting whether a reaction will occur spontaneously if the reactants and products are mixed under standard conditions. We should note, however, that very few reactions are actually carried out under standard conditions, and calculated values of ΔG° may not tell us whether a given reaction will occur spontaneously under nonstandard conditions. What determines whether a reaction will occur spontaneously is the free-energy change (ΔG) under the actual experimental conditions, which are usually different from ΔG°. If the ΔH and TΔS terms for a reaction have the same sign, for example, then it may be possible to reverse the sign of ΔG by changing the temperature, thereby converting a reaction that is not thermodynamically spontaneous, having Keq < 1, to one that is, having a Keq > 1, or vice versa. Because ΔH and ΔS usually do not vary greatly with temperature in the absence of a phase change, we can use tabulated values of ΔH° and ΔS° to calculate ΔG° at various temperatures, as long as no phase change occurs over the temperature range being considered.
In the absence of a phase change, neither \(ΔH\) nor \(ΔS\) vary greatly with temperature.
Calculate (a) ΔG° and (b) ΔG300°C for the reaction N2(g)+3H2(g)⇌2NH3(g), assuming that ΔH and ΔS do not change between 25°C and 300°C. Use these data:
- S°(N2) = 191.6 J/(mol•K),
- S°(H2) = 130.7 J/(mol•K),
- S°(NH3) = 192.8 J/(mol•K), and
- ΔH∘f (NH3) = −45.9 kJ/mol.
Given: balanced chemical equation, temperatures, S° values, and ΔH∘f for NH3
Asked for: ΔG° and ΔG at 300°C
Strategy:
- Convert each temperature to kelvins. Then calculate ΔS° for the reaction. Calculate ΔH° for the reaction, recalling that ΔH∘f for any element in its standard state is zero.
- Substitute the appropriate values into Equation \(\ref{Eq5}\) to obtain ΔG° for the reaction.
- Assuming that ΔH and ΔS are independent of temperature, substitute values into Equation \(\ref{Eq2}\) to obtain ΔG for the reaction at 300°C.
Solution
A To calculate ΔG° for the reaction using Equation \(\ref{Eq5}\), we must know the temperature as well as the values of ΔS° and ΔH°. At standard conditions, the temperature is 25°C, or 298 K. We can calculate ΔS° for the reaction from the absolute molar entropy values given for the reactants and the products using the “products minus reactants” rule:
\nonumber\\ &=[\textrm{2 mol NH}_3\times192.8\;\mathrm{J/(mol\cdot K)}]
\nonumber\\ &-\left \{[\textrm{1 mol N}_2\times191.6\;\mathrm{J/(mol\cdot K)}]+[\textrm{3 mol H}_2\times130.7\;\mathrm{J/(mol\cdot K)}]\right \} \nonumber\\ &=-198.1\textrm{ J/K (per mole of N}_2)\end{align} \nonumber\]
We can also calculate ΔH° for the reaction using the “products minus reactants” rule. The value of ΔH∘f (NH3) is given, and ΔH∘f is zero for both N2 and H2:
\[\begin{align}\Delta H^\circ_{\textrm{rxn}}&=2\Delta H^\circ_\textrm f(\mathrm{NH_3})-[\Delta H^\circ_\textrm f(\mathrm{N_2})+3\Delta H^\circ_\textrm f(\mathrm{H_2})] \nonumber \\ &=[2\times(-45.9\textrm{ kJ/mol})]-[(1\times0\textrm{ kJ/mol})+(3\times0 \textrm{ kJ/mol})] \nonumber \\ &=-91.8\textrm{ kJ(per mole of N}_2) \nonumber\end{align} \nonumber\]
B Inserting the appropriate values into Equation \(\ref{Eq5}\)
\[\Delta G^\circ_{\textrm{rxn}}=\Delta H^\circ-T\Delta S^\circ=(-\textrm{91.8 kJ})-(\textrm{298 K})(-\textrm{198.1 J/K})(\textrm{1 kJ/1000 J})=-\textrm{32.7 kJ (per mole of N}_2) \nonumber\]
C To calculate ΔG for this reaction at 300°C, we assume that ΔH and ΔS are independent of temperature (i.e., ΔH300°C = H° and ΔS300°C = ΔS°) and insert the appropriate temperature (573 K) into Equation \(\ref{Eq2}\):
\[\begin{align}\Delta G_{300^\circ\textrm C}&=\Delta H_{300^\circ\textrm C}-(\textrm{573 K})(\Delta S_{300^\circ\textrm C})=\Delta H^\circ -(\textrm{573 K})\Delta S^\circ \nonumber \\ &=(-\textrm{91.8 kJ})-(\textrm{573 K})(-\textrm{198.1 J/K})(\textrm{1 kJ/1000 J})=21.7\textrm{ kJ (per mole of N}_2) \nonumber \end{align} \nonumber \]
In this example, changing the temperature has a major effect on the thermodynamic spontaneity of the reaction. Under standard conditions, the reaction of nitrogen and hydrogen gas to produce ammonia is thermodynamically spontaneous, but in practice, it is too slow to be useful industrially. Increasing the temperature in an attempt to make this reaction occur more rapidly also changes the thermodynamics by causing the −TΔS° term to dominate, and the reaction is no longer spontaneous at high temperatures; that is, its Keq is less than one. This is a classic example of the conflict encountered in real systems between thermodynamics and kinetics, which is often unavoidable.
Calculate
- \(ΔG°\) and
- \(ΔG_{750°C}\)
for the following reaction
\[2NO_{(g)}+O_{2\; (g)} \rightleftharpoons 2NO_{2\; (g)} \nonumber\]
which is important in the formation of urban smog. Assume that \(ΔH\) and \(ΔS\) do not change between 25.0°C and 750°C and use these data:
- S°(NO) = 210.8 J/(mol•K),
- S°(O2) = 205.2 J/(mol•K),
- S°(NO2) = 240.1 J/(mol•K),
- ΔH∘f(NO2) = 33.2 kJ/mol, and
- ΔH∘f (NO) = 91.3 kJ/mol.
Answer
- −72.5 kJ/mol of \(O_2\)
- 33.8 kJ/mol of \(O_2\)
The effect of temperature on the spontaneity of a reaction, which is an important factor in the design of an experiment or an industrial process, depends on the sign and magnitude of both ΔH° and ΔS°. The temperature at which a given reaction is at equilibrium can be calculated by setting ΔG° = 0 in Equation \(\ref{Eq5}\), as illustrated in Example \(\PageIndex{4}\).
The reaction of nitrogen and hydrogen gas to produce ammonia is one in which ΔH° and ΔS° are both negative. Such reactions are predicted to be thermodynamically spontaneous at low temperatures but nonspontaneous at high temperatures. Use the data in Example \(\PageIndex{3}\) to calculate the temperature at which this reaction changes from spontaneous to nonspontaneous, assuming that ΔH° and ΔS° are independent of temperature.
Given: ΔH° and ΔS°
Asked for: temperature at which reaction changes from spontaneous to nonspontaneous
Strategy:
Set ΔG° equal to zero in Equation \(\ref{Eq5}\) and solve for T, the temperature at which the reaction becomes nonspontaneous.
Solution
In Example \(\PageIndex{3}\), we calculated that ΔH° is −91.8 kJ/mol of N2 and ΔS° is −198.1 J/K per mole of N2, corresponding to ΔG° = −32.7 kJ/mol of N2 at 25°C. Thus the reaction is indeed spontaneous at low temperatures, as expected based on the signs of ΔH° and ΔS°. The temperature at which the reaction becomes nonspontaneous is found by setting ΔG° equal to zero and rearranging Equation \(\ref{Eq5}\) to solve for T:
\\ \Delta H^\circ &=T\Delta S^\circ
\\ T=\dfrac{\Delta H^\circ}{\Delta S^\circ}&=\dfrac{(-\textrm{91.8 kJ})(\textrm{1000 J/kJ})}{-\textrm{198.1 J/K}}=\textrm{463 K}\end{align}\]
This is a case in which a chemical engineer is severely limited by thermodynamics. Any attempt to increase the rate of reaction of nitrogen with hydrogen by increasing the temperature will cause reactants to be favored over products above 463 K.
ΔH° and ΔS° are both negative for the reaction of nitric oxide and oxygen to form nitrogen dioxide. Use those data to calculate the temperature at which this reaction changes from spontaneous to nonspontaneous.
Answer: 792.6 K
Summary
- The change in Gibbs free energy, which is based solely on changes in state functions, is the criterion for predicting the spontaneity of a reaction.
We can predict whether a reaction will occur spontaneously by combining the entropy, enthalpy, and temperature of a system in a new state function called Gibbs free energy (G). The change in free energy (ΔG) is the difference between the heat released during a process and the heat released for the same process occurring in a reversible manner. If a system is at equilibrium, ΔG = 0. If the process is spontaneous, ΔG < 0. If the process is not spontaneous as written but is spontaneous in the reverse direction, ΔG > 0. At constant temperature and pressure, ΔG is equal to the maximum amount of work a system can perform on its surroundings while undergoing a spontaneous change. The standard free-energy change (ΔG°) is the change in free energy when one substance or a set of substances in their standard states is converted to one or more other substances, also in their standard states. The standard free energy of formation (ΔG∘f), is the change in free energy that occurs when 1 mol of a substance in its standard state is formed from the component elements in their standard states. Tabulated values of standard free energies of formation are used to calculate ΔG° for a reaction.