
18.3: Evaluating Entropy and Entropy Changes
- Page ID
- 46235
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)- To use thermodynamic cycles to calculate changes in entropy.
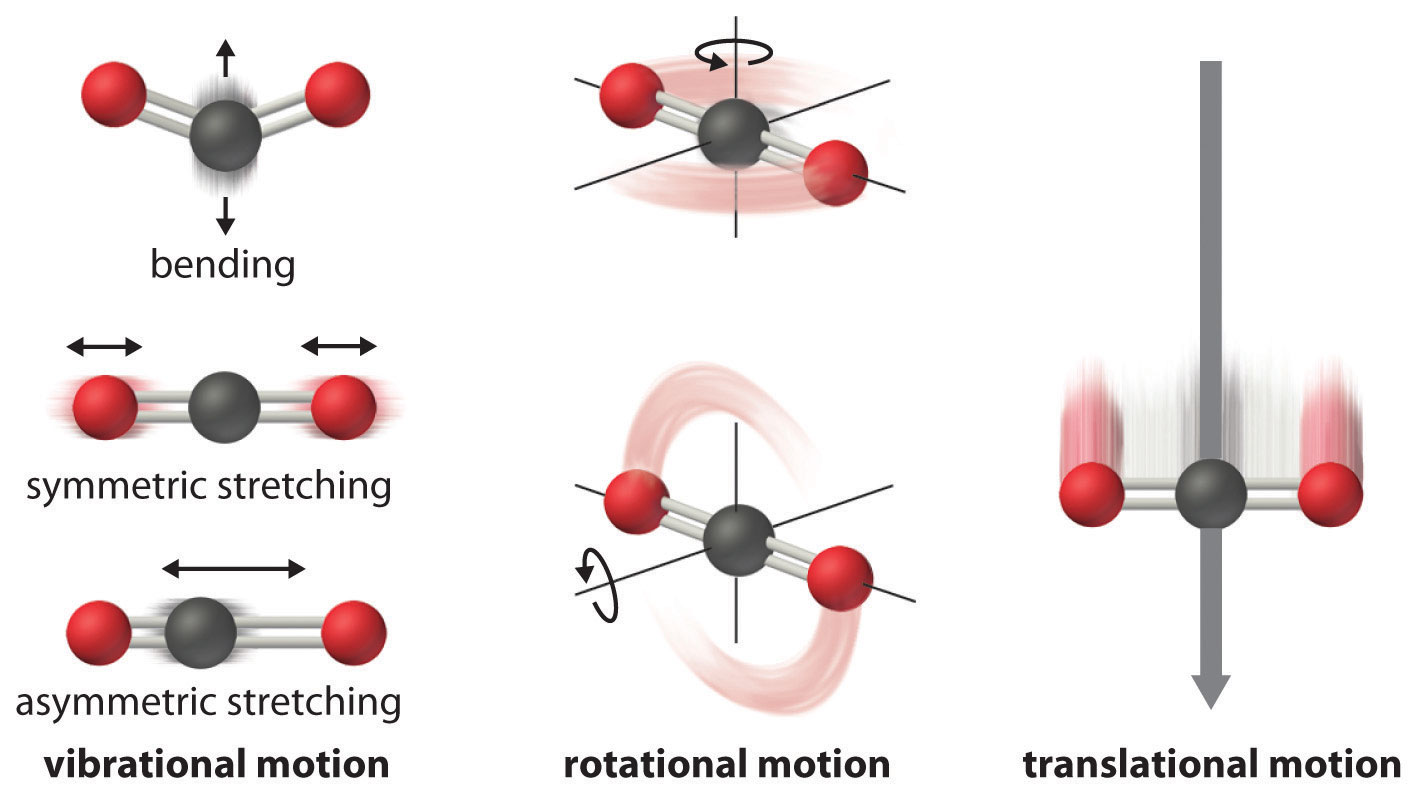
The atoms, molecules, or ions that compose a chemical system can undergo several types of molecular motion, including translation, rotation, and vibration (Figure \(\PageIndex{1}\)). The greater the molecular motion of a system, the greater the number of possible microstates and the higher the entropy. A perfectly ordered system with only a single microstate available to it would have an entropy of zero. The only system that meets this criterion is a perfect crystal at a temperature of absolute zero (0 K), in which each component atom, molecule, or ion is fixed in place within a crystal lattice and exhibits no motion (ignoring quantum effects). Such a state of perfect order (or, conversely, zero disorder) corresponds to zero entropy. In practice, absolute zero is an ideal temperature that is unobtainable, and a perfect single crystal is also an ideal that cannot be achieved. Nonetheless, the combination of these two ideals constitutes the basis for the third law of thermodynamics: the entropy of any perfectly ordered, crystalline substance at absolute zero is zero.
The entropy of any perfectly ordered, crystalline substance at absolute zero is zero.
The third law of thermodynamics has two important consequences: it defines the sign of the entropy of any substance at temperatures above absolute zero as positive, and it provides a fixed reference point that allows us to measure the absolute entropy of any substance at any temperature.In practice, chemists determine the absolute entropy of a substance by measuring the molar heat capacity (Cp) as a function of temperature and then plotting the quantity Cp/T versus T. The area under the curve between 0 K and any temperature T is the absolute entropy of the substance at T. In contrast, other thermodynamic properties, such as internal energy and enthalpy, can be evaluated in only relative terms, not absolute terms. In this section, we examine two different ways to calculate ΔS for a reaction or a physical change. The first, based on the definition of absolute entropy provided by the third law of thermodynamics, uses tabulated values of absolute entropies of substances. The second, based on the fact that entropy is a state function, uses a thermodynamic cycle similar to those discussed previously.
Calculating ΔS from Standard Molar Entropy Values
One way of calculating ΔS for a reaction is to use tabulated values of the standard molar entropy (S°), which is the entropy of 1 mol of a substance at a standard temperature of 298 K; the units of S° are J/(mol•K). Unlike enthalpy or internal energy, it is possible to obtain absolute entropy values by measuring the entropy change that occurs between the reference point of 0 K [corresponding to S = 0 J/(mol•K)] and 298 K.
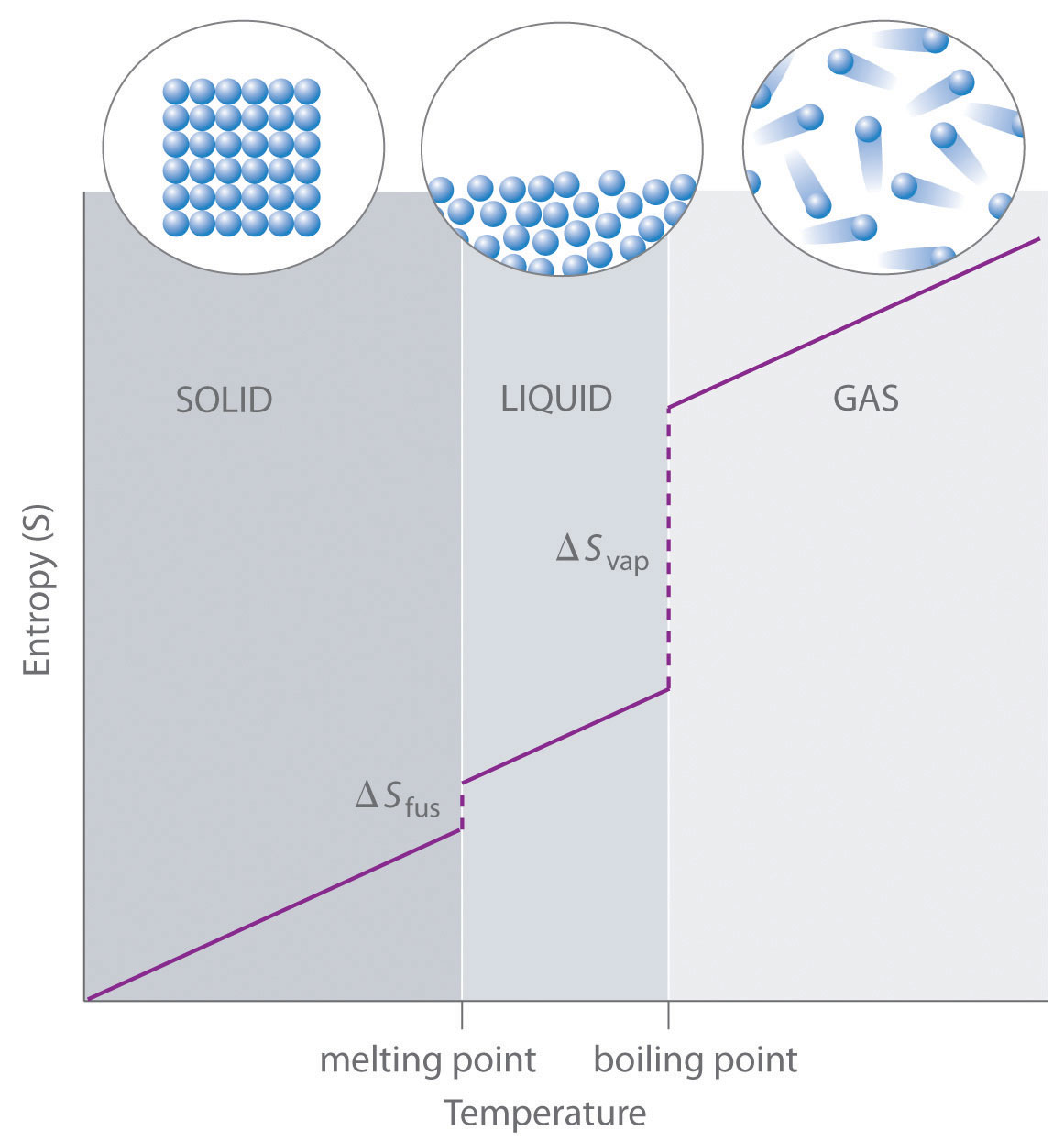
As shown in Table \(\PageIndex{1}\), for substances with approximately the same molar mass and number of atoms, S° values fall in the order S°(gas) > S°(liquid) > S°(solid). For instance, S° for liquid water is 70.0 J/(mol•K), whereas S° for water vapor is 188.8 J/(mol•K). Likewise, S° is 260.7 J/(mol•K) for gaseous I2 and 116.1 J/(mol•K) for solid I2. This order makes qualitative sense based on the kinds and extents of motion available to atoms and molecules in the three phases. The correlation between physical state and absolute entropy is illustrated in Figure \(\PageIndex{2}\), which is a generalized plot of the entropy of a substance versus temperature.
| Gases | Liquids | Solids | |||
| Substance | S° [J/(mol•K)] | Substance | S° [J/(mol•K)] | Substance | S° [J/(mol•K)] |
| He | 126.2 | H2O | 70.0 | C (diamond) | 2.4 |
| H2 | 130.7 | CH3OH | 126.8 | C (graphite) | 5.7 |
| Ne | 146.3 | Br2 | 152.2 | LiF | 35.7 |
| Ar | 154.8 | CH3CH2OH | 160.7 | SiO2 (quartz) | 41.5 |
| Kr | 164.1 | C6H6 | 173.4 | Ca | 41.6 |
| Xe | 169.7 | CH3COCl | 200.8 | Na | 51.3 |
| H2O | 188.8 | C6H12 (cyclohexane) | 204.4 | MgF2 | 57.2 |
| N2 | 191.6 | C8H18 (isooctane) | 329.3 | K | 64.7 |
| O2 | 205.2 | NaCl | 72.1 | ||
| CO2 | 213.8 | KCl | 82.6 | ||
| I2 | 260.7 | I2 | 116.1 | ||
Entropy increases with softer, less rigid solids, solids that contain larger atoms, and solids with complex molecular structures.
A closer examination of Table \(\PageIndex{1}\) also reveals that substances with similar molecular structures tend to have similar S° values. Among crystalline materials, those with the lowest entropies tend to be rigid crystals composed of small atoms linked by strong, highly directional bonds, such as diamond [S° = 2.4 J/(mol•K)]. In contrast, graphite, the softer, less rigid allotrope of carbon, has a higher S° [5.7 J/(mol•K)] due to more disorder in the crystal. Soft crystalline substances and those with larger atoms tend to have higher entropies because of increased molecular motion and disorder. Similarly, the absolute entropy of a substance tends to increase with increasing molecular complexity because the number of available microstates increases with molecular complexity. For example, compare the S° values for CH3OH(l) and CH3CH2OH(l). Finally, substances with strong hydrogen bonds have lower values of S°, which reflects a more ordered structure.
ΔS° for a reaction can be calculated from absolute entropy values using the same “products minus reactants” rule used to calculate ΔH°.
To calculate ΔS° for a chemical reaction from standard molar entropies, we use the familiar “products minus reactants” rule, in which the absolute entropy of each reactant and product is multiplied by its stoichiometric coefficient in the balanced chemical equation. Example \(\PageIndex{1}\) illustrates this procedure for the combustion of the liquid hydrocarbon isooctane (C8H18; 2,2,4-trimethylpentane).
Use the data in Table \(\PageIndex{1}\) to calculate ΔS° for the reaction of liquid isooctane with O2(g) to give CO2(g) and H2O(g) at 298 K.
Given: standard molar entropies, reactants, and products
Asked for: ΔS°
Strategy:
Write the balanced chemical equation for the reaction and identify the appropriate quantities in Table \(\PageIndex{1}\). Subtract the sum of the absolute entropies of the reactants from the sum of the absolute entropies of the products, each multiplied by their appropriate stoichiometric coefficients, to obtain ΔS° for the reaction.
Solution:
The balanced chemical equation for the complete combustion of isooctane (C8H18) is as follows:
We calculate ΔS° for the reaction using the “products minus reactants” rule, where m and n are the stoichiometric coefficients of each product and each reactant:
\begin{align}\Delta S^\circ_{\textrm{rxn}}&=\sum mS^\circ(\textrm{products})-\sum nS^\circ(\textrm{reactants})
\\ &=[8S^\circ(\mathrm{CO_2})+9S^\circ(\mathrm{H_2O})]-[S^\circ(\mathrm{C_8H_{18}})+\dfrac{25}{2}S^\circ(\mathrm{O_2})]
\\ &=\left \{ [8\textrm{ mol }\mathrm{CO_2}\times213.8\;\mathrm{J/(mol\cdot K)}]+[9\textrm{ mol }\mathrm{H_2O}\times188.8\;\mathrm{J/(mol\cdot K)}] \right \}
\\ &-\left \{[1\textrm{ mol }\mathrm{C_8H_{18}}\times329.3\;\mathrm{J/(mol\cdot K)}]+\left [\dfrac{25}{2}\textrm{ mol }\mathrm{O_2}\times205.2\textrm{ J}/(\mathrm{mol\cdot K})\right ] \right \}
\\ &=515.3\;\mathrm{J/K}\end{align}
ΔS° is positive, as expected for a combustion reaction in which one large hydrocarbon molecule is converted to many molecules of gaseous products.
Use the data in Table \(\PageIndex{1}\) to calculate ΔS° for the reaction of H2(g) with liquid benzene (C6H6) to give cyclohexane (C6H12).
Answer: −361.1 J/K
Calculating ΔS from Thermodynamic Cycles
We can also calculate a change in entropy using a thermodynamic cycle. As you learned previously, the molar heat capacity (Cp) is the amount of heat needed to raise the temperature of 1 mol of a substance by 1°C at constant pressure. Similarly, Cv is the amount of heat needed to raise the temperature of 1 mol of a substance by 1°C at constant volume. The increase in entropy with increasing temperature in Figure \(\PageIndex{2}\) is approximately proportional to the heat capacity of the substance.
Recall that the entropy change (ΔS) is related to heat flow (qrev) by ΔS = qrev/T. Because qrev = nCpΔT at constant pressure or nCvΔT at constant volume, where n is the number of moles of substance present, the change in entropy for a substance whose temperature changes from T1 to T2 is as follows:
\[\Delta S=\dfrac{q_{\textrm{rev}}}{T}=nC_\textrm p\dfrac{\Delta T}{T}\hspace{4mm}(\textrm{constant pressure})\]
As you will discover in more advanced math courses than is required here, it can be shown that this is equal to the following:For a review of natural logarithms, see Essential Skills 6 in Chapter 11.
\[\Delta S=nC_\textrm p\ln\dfrac{T_2}{T_1}\hspace{4mm}(\textrm{constant pressure}) \label{18.20}\]
Similarly,
\[\Delta S=nC_{\textrm v}\ln\dfrac{T_2}{T_1}\hspace{4mm}(\textrm{constant volume}) \label{18.21}\]
Thus we can use a combination of heat capacity measurements (Equation 18.20 or Equation 18.21) and experimentally measured values of enthalpies of fusion or vaporization if a phase change is involved (Equation 18.18) to calculate the entropy change corresponding to a change in the temperature of a sample.
We can use a thermodynamic cycle to calculate the entropy change when the phase change for a substance such as sulfur cannot be measured directly. As noted in the exercise in Example 6, elemental sulfur exists in two forms (part (a) in Figure \(\PageIndex{3}\)): an orthorhombic form with a highly ordered structure (Sα) and a less-ordered monoclinic form (Sβ). The orthorhombic (α) form is more stable at room temperature but undergoes a phase transition to the monoclinic (β) form at temperatures greater than 95.3°C (368.5 K). The transition from Sα to Sβ can be described by the thermodynamic cycle shown in part (b) in Figure \(\PageIndex{3}\), in which liquid sulfur is an intermediate. The change in entropy that accompanies the conversion of liquid sulfur to Sβ (−ΔSfus(β) = ΔS3 in the cycle) cannot be measured directly. Because entropy is a state function, however, ΔS3 can be calculated from the overall entropy change (ΔSt) for the Sα–Sβ transition, which equals the sum of the ΔS values for the steps in the thermodynamic cycle, using Equation 18.20 and tabulated thermodynamic parameters (the heat capacities of Sα and Sβ, ΔHfus(α), and the melting point of Sα.)

If we know the melting point of Sα (Tm = 115.2°C = 388.4 K) and ΔSt for the overall phase transition [calculated to be 1.09 J/(mol•K) in the exercise in Example 6], we can calculate ΔS3 from the values given in part (b) in Figure \(\PageIndex{3}\) where Cp(α) = 22.70 J/mol•K and Cp(β) = 24.77 J/mol•K (subscripts on ΔS refer to steps in the cycle):
\(\begin{align}\Delta S_{\textrm t}&=\Delta S_1+\Delta S_2+\Delta S_3+\Delta S_4
\\ 1.09\;\mathrm{J/(mol\cdot K)}&=C_{\textrm p({\alpha})}\ln\left(\dfrac{T_2}{T_1}\right)+\dfrac{\Delta H_{\textrm{fus}}}{T_{\textrm m}}+\Delta S_3+C_{\textrm p(\beta)}\ln\left(\dfrac{T_4}{T_3}\right)
\\ &=22.70\;\mathrm{J/(mol\cdot K)}\ln\left(\dfrac{388.4}{368.5}\right)+\left(\dfrac{1.722\;\mathrm{kJ/mol}}{\textrm{388.4 K}}\times1000\textrm{ J/kJ}\right)
\\ &+\Delta S_3+24.77\;\mathrm{J/(mol\cdot K)}\ln\left(\dfrac{368.5}{388.4}\right)
\\ &=[1.194\;\mathrm{J/(mol\cdot K)}]+[4.434\;\mathrm{J/(mol\cdot K)}]+\Delta S_3+[-1.303\;\mathrm{J/(mol\cdot K)}]\end{align}\)
Solving for ΔS3 gives a value of −3.24 J/(mol•K). As expected for the conversion of a less ordered state (a liquid) to a more ordered one (a crystal), ΔS3 is negative.
The absolute entropy of a substance at any temperature above 0 K must be determined by calculating the increments of heat q required to bring the substance from 0 K to the temperature of interest, and then summing the ratios q/T. Two kinds of experimental measurements are needed:
- The enthalpies associated with any phase changes the substance may undergo within the temperature range of interest. Melting of a solid and vaporization of a liquid correspond to sizeable increases in the number of microstates available to accept thermal energy, so as these processes occur, energy will flow into a system, filling these new microstates to the extent required to maintain a constant temperature (the freezing or boiling point); these inflows of thermal energy correspond to the heats of fusion and vaporization. The entropy increase associated with melting, for example, is just ΔHfusion/Tm.
- The heat capacity C of a phase expresses the quantity of heat required to change the temperature by a small amount ΔT , or more precisely, by an infinitesimal amount dT . Thus the entropy increase brought about by warming a substance over a range of temperatures that does not encompass a phase transition is given by the sum of the quantities C dT/T for each increment of temperature dT . This is of course just the integral
\[ S_{0 \rightarrow T^o} = \int _{0}^{T^o} \dfrac{C_p}{T} dt \]
Because the heat capacity is itself slightly temperature dependent, the most precise determinations of absolute entropies require that the functional dependence of C on T be used in the above integral in place of a constant C.
\[ S_{0 \rightarrow T^o} = \int _{0}^{T^o} \dfrac{C_p(T)}{T} dt \]
When this is not known, one can take a series of heat capacity measurements over narrow temperature increments ΔT and measure the area under each section of the curve.
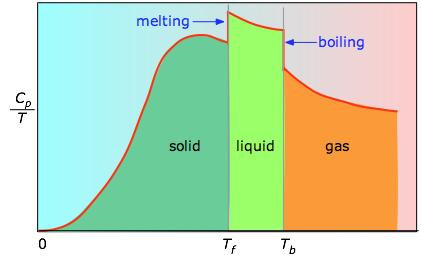
The area under each section of the plot represents the entropy change associated with heating the substance through an interval ΔT. To this must be added the enthalpies of melting, vaporization, and of any solid-solid phase changes. Values of Cp for temperatures near zero are not measured directly, but can be estimated from quantum theory.
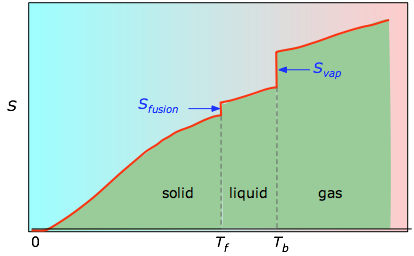
The cumulative areas from 0 K to any given temperature (taken from the experimental plot on the left) are then plotted as a function of T, and any phase-change entropies such as Svap = Hvap / Tb are added to obtain the absolute entropy at temperature T.
Summary
- Entropy changes can be calculated using the “products minus reactants” rule or from a combination of heat capacity measurements and measured values of enthalpies of fusion or vaporization.
The third law of thermodynamics states that the entropy of any perfectly ordered, crystalline substance at absolute zero is zero. At temperatures greater than absolute zero, entropy has a positive value, which allows us to measure the absolute entropy of a substance. Measurements of the heat capacity of a substance and the enthalpies of fusion or vaporization can be used to calculate the changes in entropy that accompany a physical change. The entropy of 1 mol of a substance at a standard temperature of 298 K is its standard molar entropy (S°). We can use the “products minus reactants” rule to calculate the standard entropy change (ΔS°) for a reaction using tabulated values of S° for the reactants and the products.