
7.3.4: Wacker (Smidt) Process
- Page ID
- 429335

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)See also Wacker Oxidation (Wikipedia)
The Wacker oxidation refers generally to the transformation of a terminal or 1,2-disubstituted alkene to a ketone through the action of catalytic palladium(II), water, and a co-oxidant. Variants of the reaction yield aldehydes, allylic/vinylic ethers, and allylic/vinylic amines. Because of the ease with which terminal alkenes may be prepared and the versatility of the methyl ketone group installed by the reaction, the Wacker oxidation has been employed extensively in organic synthesis.[1]
Introduction
The stoichiometric conversion of ethylene to acetaldehyde by an acidic, aqueous solution of PdCl2 was discovered over a century ago,[2] but fifty years passed between the discovery of this reaction and the development of a catalytic method. In 1959, researchers at Wacker Chemie reported that a similar transformation takes place in an aqueous, acidic solution of catalytic PdCl2 and a stoichiometric amount of CuCl2 through which oxygen is bubbled (Equ. \(\PageIndex{1}\)).[3]
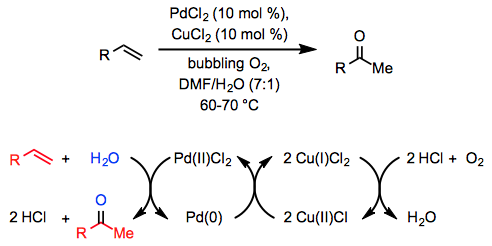
Since this initial report, the Wacker process has been widely applied in organic synthesis and has been extended to other classes of substrates and products. To encourage mixing of the organic reactants with the aqueous phase, a co-solvent is generally employed along with water. Dimethylformamide (DMF) is a common choice; when DMF is used as a co-solvent with a stoichiometric amount of CuCl under balloon pressure of oxygen, the reaction is called the "Tsuji-Wacker oxidation."[4] Applications of the Wacker oxidation to organic synthesis typically involve the installation of a methyl ketone moiety, which may subsequently undergo nucleophilic addition or deprotonation to form an enolate.
Mechanism and Stereochemistry
Prevailing Mechanism: Water Nucleophile
The mechanism of the Wacker oxidation has been studied both experimentally and theoretically (Eq. 2). The first step of the Wacker oxidation involves coordination of the the alkene to the palladium center to form π-complex 2. Evidence for this step is provided by the relative sluggishness of electron-poor alkenes, which generally require higher catalyst loadings than unactivated alkenes. Hydroxypalladation then occurs to yield either zwitterionic complex 3 or neutral complex 4 depending on the mode of hydroxypalladation (see below). Studies employing deuterated substrates suggest that β-hydride elimination then occurs to afford enol complex 5, which re-inserts into the Pd-H bond to afford complex 6.[5] Computational studies support the involvement of chloride-assisted deprotonation in the subsequent step,[6] which affords the product and palladium(0). Oxidation of palladium(0) by copper(II) then occurs, regenerating palladium(II) species 1. The role of copper(II) in the mechanism is poorly understood at present.
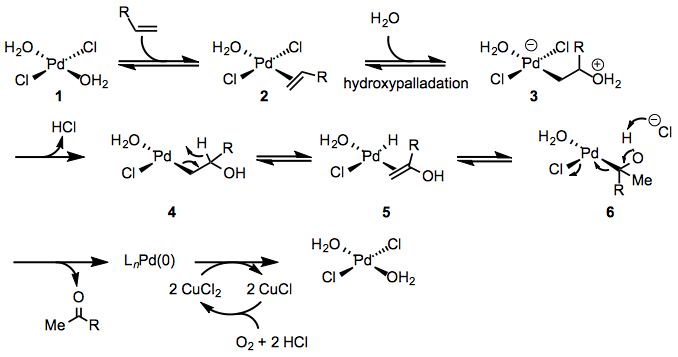
(2)
The mode of hydroxypalladation is an important issue for the Wacker oxidation. Hydroxypalladation may occur either in a syn fashion through an inner-sphere mechanism or in an anti fashion via nucleophilic attack on the coordinated alkene (Eq. 3). Although the stereocenter in 4 is ultimately destroyed upon elimination, the mode of hydroxypalladation can influence the site selectivity of the reaction. Markovnikov-type addition of water to the more substituted carbon of the alkene forms a methyl ketone, while attack of water at the less substituted position ultimately yields an aldehyde. The mode of hydroxypalladation has been shown to affect the distribution of ketone and aldehyde products, and is also important in stereoselective Wacker cyclization reactions.
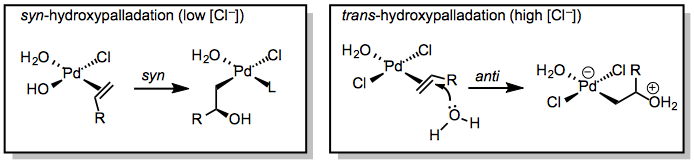
(3)
At low concentrations of chloride ion, syn-hydroxypalladation appears to be the norm.[7] Water coordinates to the metal and migratory insertion into the Pd-OH bond occurs to generate 4 directly. At high concentrations of chloride ion, chloride competes with hydroxide for binding at palladium, and thus anti-hydroxypalladation may occur.[8]
Prevailing Mechanism: Alcohol Nucleophiles
Alcohols may also be employed as nucleophiles in the Wacker process. The initial steps of the mechanism are similar to those for the Wacker oxidation with water, but the mechanisms diverge at alkoxy complex 11 (Eq. 4). Elimination of palladium occurs to generate an oxocarbenium ion, which may be captured by solvent to generate an acetal. Small amounts of vinyl ether products support the intermediacy of complex 10.[9]
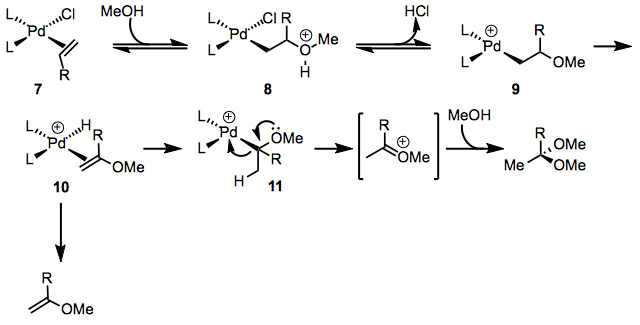
Enantioselective Variants
When the substrate contains a tethered nucleophile, such as a hydroxyl group or protected amine, the nucleophile may react to form a cyclic, allylic or vinylic ether or amine after β-hydride elimination. This process is known as the "Wacker cyclization," and when the product is allylic, a stereocenter may be established. Chiral ligands have been used to render the Wacker cyclization enantioselective. For example, use of tetra(dihydrooxazoline) ligand 12 and benzoquinone (BQ) as a co-oxidant resulted in formation of allylic ether product in an enantiomeric ratio of 98:2 (Eq. 5). [10]
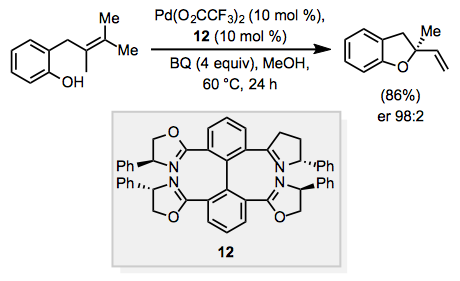 Equation \(\PageIndex{5}\)
Equation \(\PageIndex{5}\)The Wacker process may also establish a stereocenter in 1,1-disubstituted alkenes via acetal formation. A chiral auxiliary in the alkene has been used successfully to generate chiral acetals with high diastereomeric ratio. The acetal forms at the more electron-deficient site of the alkene (Eq. 6).[11]
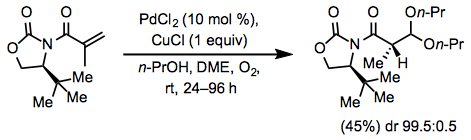
Scope and Limitations
Terminal alkenes are the most commonly employed substrates for the Wacker oxidation. Oxygen-containing functionality far from the reactive alkene is well tolerated by the reaction because palladium reacts selectively with the alkene in preference to lone pairs on oxygen. Wacker oxidation followed by intramolecular aldol condensation is a convenient method for the rapid synthesis of carbocycles (Eq. 7).[12]
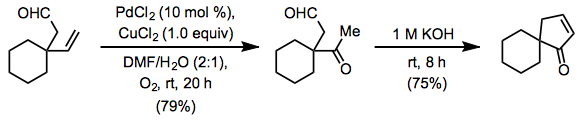
The original Wacker process and Tsuji-Wacker conditions can present problems for substrates containing acid-sensitive functionality, such as acetals and silyl ethers. Use of copper(II) chloride leads to the generation of strongly acidic hydrochloric acid. To mitigate this issue, a method employing copper(II) acetate has been developed. Some acid-sensitive groups are able to withstand the milder acetic acid generated by this method (Eq. 8).[13] Under similar conditions involving copper(I) chloride, a yield of only 56% was obtained.
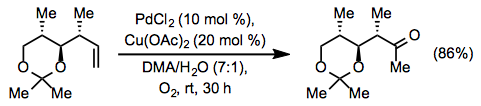
Unprotected amines coordinate strongly to palladium and thus cause problems for Wacker oxidations. Amines protected with electron-withdrawing substituents often do not interfere in Wacker oxidations,[14] although they may participate in aza-Wacker cyclizations if appropriately positioned in the substrate (see below).
Tri-substituted alkenes are completely inert to the conditions of the Wacker oxidation; only terminal and 1,1-disubstituted alkenes react. Terminal alkenes are usually oxidized selectively over internal alkenes, except under rather specialized circumstances.[15] Eq. 9 represents a typical case of selectivity for terminal alkenes.[14]
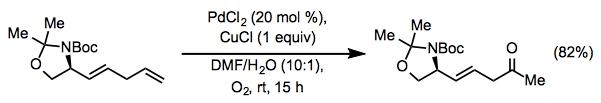
Lewis-basic functionality in the allylic or homoallylic position relative to the reactive alkene may cause unpredictable site selectivity in Wacker oxidations. Under standard Tsuji-Wacker conditions, unprotected allylic alcohols form mixtures of methyl ketone and aldehyde products.[16] The catalyst Pd(Quinox)Cl2 was developed to address this problem; allylic alcohols react selectively in the presence of this catalyst to afford methyl ketones (Eq. 10).[17] tert-Butyl hydroperoxide (TBHP) is used as the oxygen source in this reaction.
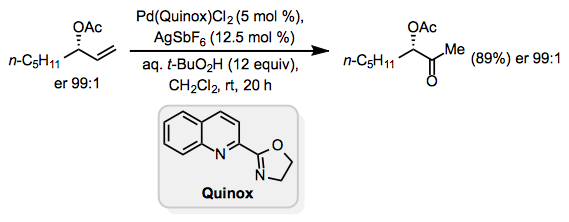 Equation \(\PageIndex{10}\)
Equation \(\PageIndex{10}\)Protected, homoallylic alcohols generally form the methyl ketone selectively, but results are more unpredictable for unprotected homoallylic alcohols.[18]
Protected allylic amines generally give higher yields of aldehyde than comparable protected allylic alcohols; these results suggest that coordination of palladium to the Lewis base is responsible for the formation of aldehydes. Using a phthalimide group in the allylic position, the Wacker oxidation may be rendered completely selective for aldehydes (Eq. 11).[19]
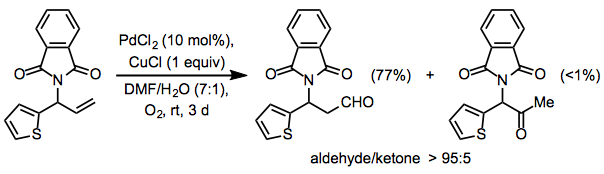 Equation \(\PageIndex{11}\)
Equation \(\PageIndex{11}\)Site selectivity in reactions of internal alkenes is low unless an allylic or homoallylic Lewis base is present in the substrate. Electron-withdrawing groups can direct oxidation to the β position of the alkene (Eq. 12), but relatively high catalyst loadings are required.[20]
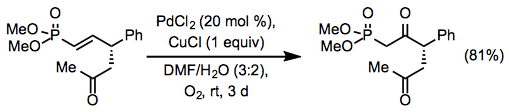 Equation \(\PageIndex{12}\)
Equation \(\PageIndex{12}\)When an appropriately situated nucleophile is present in the substrate, Wacker cyclization may occur to yield an allylic or vinylic ether (Eq. 13).[21] The aza-Wacker cyclization involves protected amino nucleophiles such as sulfonamides.[22]
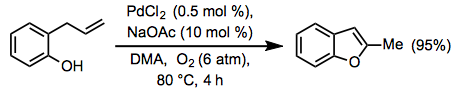 Equation \(\PageIndex{13}\)
Equation \(\PageIndex{13}\)When an alcohol is used in stoichiometric or solvent quantities or a diol is present in the substrate, acetal formation may occur. Acetal formation, like ketone formation, generally occurs with Markovnikov selectivity. For example, a Michael acceptor forms an acetal at the β position in the presence of (R,R)-2,4-pentanediol (Eq. 14).[23]
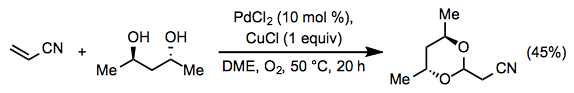 Equation \(\PageIndex{14}\)
Equation \(\PageIndex{14}\)Synthetic Applications
The Wacker oxidation has been used extensively in organic synthesis. A number of methods exist for the installation of terminal alkenes and the methyl ketone products of the reaction can be elaborated easily to more complex compounds. In addition, the reaction is aerobic and operationally simple.
The Wacker oxidation was employed in the course of synthesis of the macrolide elaiolide. Oxidation at two terminal alkenes in a C2-symmetric intermediate afforded a diketone, which was subsequently elaborated to the symmetric natural product (Eq. 15).[24] Notably, the internal alkenes and esters in the intermediate appeared not to affect the oxidation.
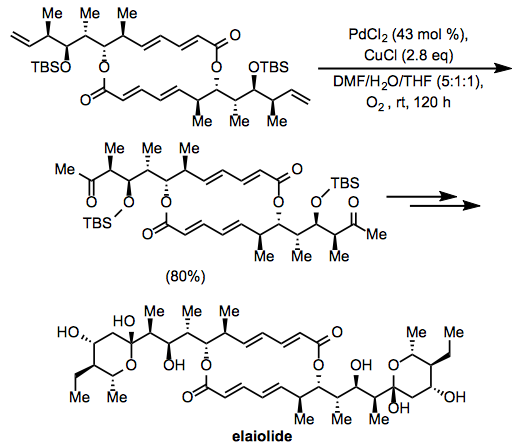 Equation \(\PageIndex{15}\)
Equation \(\PageIndex{15}\)A modification of Tsuji-Wacker conditions employing copper(II) acetate was used in the course of a synthesis of hennoxazole A (Eq. 16).[25] After the oxidation, deprotection under acidic conditions led to the intramolecular formation of an acetal present in the target compound.
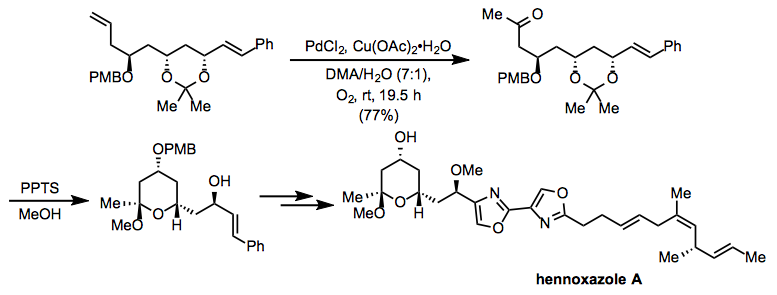 Equation \(\PageIndex{16}\)
Equation \(\PageIndex{16}\)Comparison to Other Methods
Few true alternatives to the Wacker oxidation (that is, other hydration-oxidation reactions) are known. One rather harsh method involves oxymercuration followed by transmetalation to palladium (Eq. 17).[26]
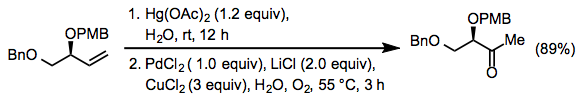 Equation \(\PageIndex{17}\)
Equation \(\PageIndex{17}\)Experimental Conditions and Procedure
Typical Conditions
Tsuji-Wacker conditions represent a good starting point for experimental conditions for this reaction. Water is mixed with an organic co-solvent (often DMF) and 10 mol% PdCl2, stoichiometric CuCl, and 1 atmosphere of oxygen gas. Copper(I) salts are generally superior to copper(II) salts because the former minimize the concentration of chloride in solution, encouraging syn-hydroxypalladation. Copper(I) is rapidly oxidized to copper(II) by dissolved oxygen; a period of 30 minutes prior to the introduction of the substrate is used to ensure that this oxidation is complete. For situations in which Tsuji-Wacker conditions fail, a wide variety of modifications of the reaction conditions may be employed.
Using high oxygen pressure and a judicious choice of solvent, Wacker oxidations can be carried out without the need for a co-oxidant. Certain ligands also promote direct O2-coupled oxidations.[27]
Peroxides may also be used as the oxygen source in Wacker oxidations; these reactions do not require a co-oxidant. In the Pd(Quinox)/TBHP system, silver hexafluoroantimonate serves as a scavenger of chloride ions.
Experimental Procedure[4]
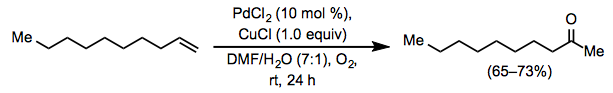 Equation \(\PageIndex{18}\)
Equation \(\PageIndex{18}\)A 100-mL, 3-necked, round-bottomed flask was fitted with a magnetic stirring bar and a pressure-equalizing dropping funnel containing 1-decene (4.2 g, 30 mmol, 1.0 equiv). The flask was charged with a mixture of PdCl2 (0.53 g, 3 mmol, 0.1 equiv), CuCl (2.97 g, 30 mmol, 1.0 equiv) and aqueous DMF (DMF/H 2O = 7:1, 24 mL). With the other outlets securely stoppered and wired down, an oxygen-filled balloon was placed over one neck, and the mixture was stirred at rt to allow oxygen uptake. After 1 h, the 1-decene (4.2 g, 30 mmol) was added over 10 min, and the solution was stirred vigorously at rt under an oxygen balloon. The color of the solution turned from green to black within 15 min and returned gradually to green. After 24 h, the mixture was poured into cold 3 N HCl (100 mL) and extracted with five, 50 mL portions of ether. The extracts were combined and washed successively with 50 mL of saturated aqueous NaHCO3 solution, 50 mL of brine, and then dried over anhydrous magnesium sulfate. The solvent was removed by evaporation and the residue was distilled using a 15 cm Vigreux column to give 2-decanone as a colorless oil (3.0–3.4 g, 65–73%): bp 43.5 °C (1 mm Hg); IR (neat) 1722 cm–1; 1H NMR (CCl4) δ 2.37 (t, J = 7 Hz, 2H), 2.02 (s, 3H), 0.7–1.8 (m, 15H).
References
- Michel, B. W.; Steffens, L. D.; Sigman, M. S. Org. React. 2014, 84, 2. (doi: 10.1002/0471264180.or084.02)
- Phillips, F. C. Am. Chem. J. 1894, 16, 255.
- Smidt, J.; Hafner, W.; Jira, R.; Sedlmeier, J.; Sieber, R.; Ruttinger, R.; Kojer, H. Angew. Chem. 1959, 71, 176.
- a b Tsuji, J.; Nagashima, H.; Nemoto, H. Org. Synth. 1984, 62, 9.
- Smidt, J.; Hafner, W.; Jira, R.; Sieber, R.; Sedlmeier, J.; Sabel, A. Angew. Chem. 1962, 74, 93.
- Eshtiagh-Hosseini, H.; Beyramabadi, S. A.; Morsali, A.; Housaindokht, M. R. J. Mol. Struct.: THEOCHEM 2010, 941, 138.
- Henry, P. M. J. Am. Chem. Soc. 1964, 86, 3246.
- Bäckvall, J. E.; Akermark, B.; Ljunggren, S. O. J. Chem. Soc., Chem. Commun. 1977, 264.
- Balija, A. M.; Stowers, K. J.; Schultz, M. J.; Sigman, M. S. Org. Lett. 2006, 8, 1121.
- Zhang, Y. J.; Wang, F.; Zhang, W. J. Org. Chem. 2007, 72, 9208.
- Hosokawa, T.; Yamanaka, T.; Itotani, M.; Murahashi, S.-I. J. Org. Chem. 1995, 60, 6159.
- Srikrishna, A.; Kumar, P. P. J. Indian Chem. Soc. 1999, 76, 521.
- Smith, A. B., III; Cho, Y. S.; Friestad, G. K. Tetrahedron Lett. 1998, 39, 8765.
- a b Reginato, G.; Mordini, A.; Verrucci, M.; Degl'Innocenti, A.; Capperucci, A. Tetrahedron: Asymmetry 2000, 11, 3759.
- Ho, T.-L.; Chang, M. H.; Chen, C. Tetrahedron Lett. 2003, 44, 6955.
- Muzart, J. Tetrahedron 2007, 63, 7505.
- Michel, B. W.; Camelio, A. M.; Cornell, C. N.; Sigman, M. S. J. Am. Chem. Soc. 2009, 131, 6076.
- Kang, S.-K.; Jung, K.-Y.; Chung, J.-U.; Namkoong, E.-Y.; Kim, T.-H. J. Org. Chem. 1995, 60, 4678.
- Weiner, B.; Baeza, A.; Jerphagnon, T.; Feringa, B. L. J. Am. Chem. Soc. 2009, 131, 9473.
- Yan, B.; Spilling, C. D. J. Org. Chem. 2008, 73, 5385.
- Mitsudome, T.; Umetani, T.; Nosaka, N.; Mori, K.; Mizugaki, T.; Ebitani, K.; Kaneda, K. Angew. Chem., Int. Ed. 2006, 45, 481.
- Hegedus, L. S.; McKearin, J. M. J. Am. Chem. Soc. 1982, 104, 2444.
- Hosokawa, T.; Ataka, Y.; Murahashi, S. Bull. Chem. Soc. Jpn. 1990, 63, 166.
- Barth, R.; Mulzer, J. Tetrahedron 2008, 64, 4718.
- Yokokawa, F.; Asano, T.; Shioiri, T. Tetrahedron 2001, 57, 6311.
- Ellis, J. M.; Crimmins, M. T. Chem. Rev. 2008, 108, 5278.
- Cornell, C. N.; Sigman, M. S. Org. Lett. 2006, 8, 4117.
Contributors
- This page is based on Organic Reactions. The primary collection of full Organic Reactions chapters is maintained by John Wiley & Sons.