
10.6: Carbocation Rearrangements
- Page ID
- 242900

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)Earlier in this chapter we introduced the so-called 'Markovnikov rule', which can be used to predict the favored regiochemical outcome of electrophilic additions to asymmetric alkenes. According to what we have learned, addition of \(HBr\) to 3-methyl-1-butene should result in a secondary bromoalkane. However, the predominant product that is actually be observed in this reaction is a tertiary alkyl bromide! Little or no secondary alkyl bromide forms.
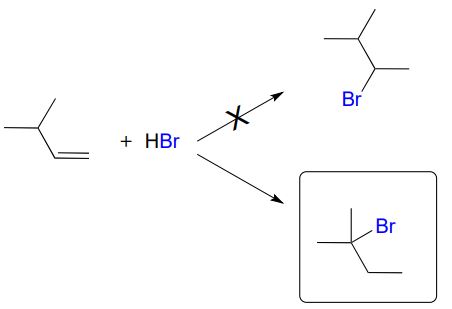
To explain this result, let's take a look at the mechanism for the reaction:
Electrophilic addition with a hydride shift:
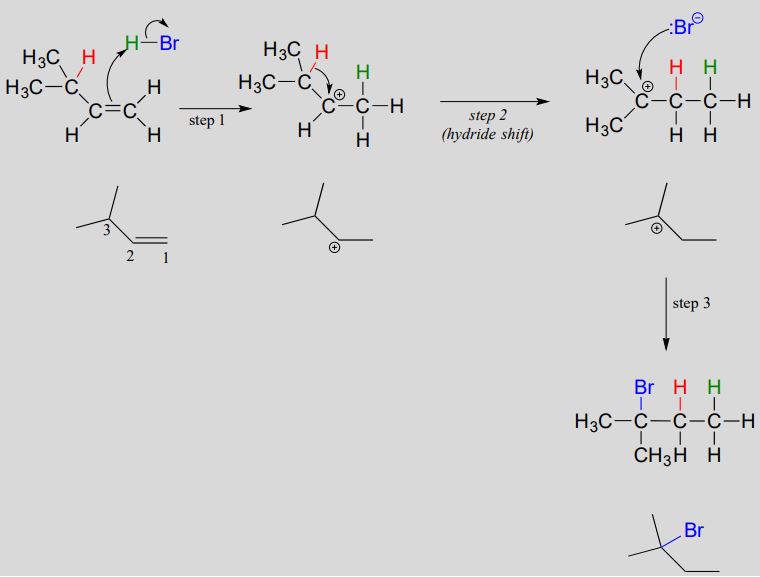
Protonation of the double bond results in a secondary carbocation (step 1). What happens next (step 2 above) is a process called a carbocation rearrangement, and more specifically, a hydride shift. The electrons in the bond between carbon #3 and a hydrogen are attracted by the positive charge on carbon #2, and they simply shift over to fill the empty \(p\) orbital, pulling the proton over with them. Notice that the hydride, in shifting, is not acting as an actual leaving group - a hydride ion is a very strong base and a very poor leaving group.
An important reminder
A hydride ion (\(H^-\)) is a proton plus two electrons. Be sure not to confuse a hydride ion with \(H^+\), which is just a proton without any electrons.
As the shift proceeds, a new \(C-H\) \(\sigma \) bond is formed at carbon #2, and carbon #3 is left with an empty \(p\) orbital and a positive charge.

What is the thermodynamic driving force for this process? Notice that the hydride shift results in the conversion of a secondary carbocation to a (more stable) tertiary carbocation - a thermodynamically downhill step. As it turns out, the shift occurs so quickly that it is accomplished before the bromide nucleophile has time to attack at carbon #2. Rather, the bromide will attack (step 3) at carbon #3 to complete the addition.
Consider another example. When \(HBr\) is added to 3,3-dimethyl-1-butene, the product is a tertiary - rather than a secondary - alkyl bromide.
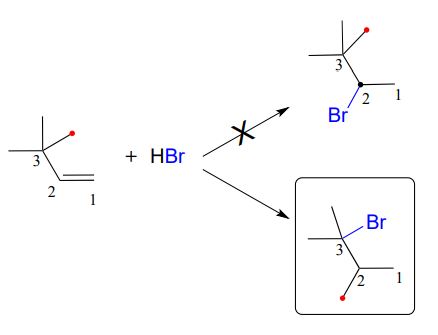
Notice that in the observed product, the carbon framework has been rearranged: the methyl carbon indicated by a red dot has shifted from carbon #3 to carbon #2. This is an example of another type of carbocation rearrangement, called a methyl shift.
Below is the mechanism for the reaction. Once again a secondary carbocation intermediate is formed in step 1. In this case, there is no hydrogen on carbon #3 available to shift over create a more stable tertiary carbocation. Instead, it is a methyl group that does the shifting, as the electrons in the carbon-carbon \(\sigma \) bond move over to fill the empty orbital on carbon #2 (step 2 below).
Electrophilic addition with methyl shift:
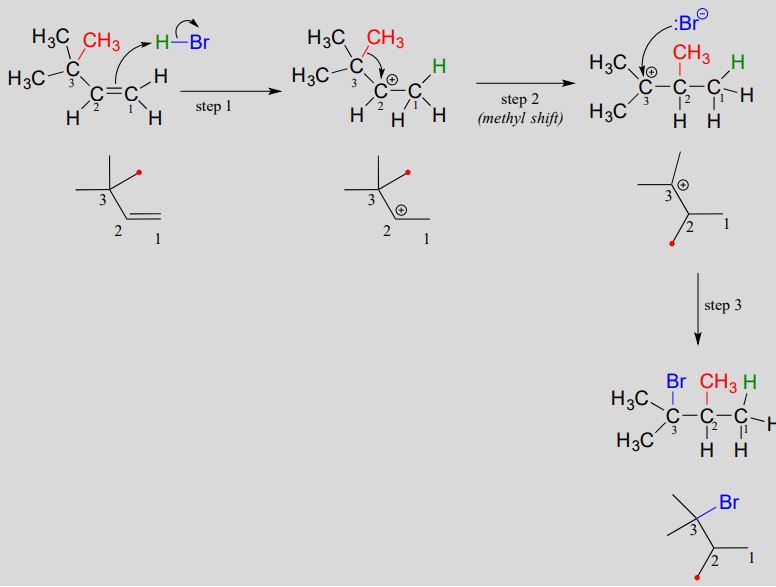
The methyl shift results in the conversion of a secondary carbocation to a more stable tertiary carbocation. The end result is a rearrangement of the carbon framework of the molecule.
Exercise \(\PageIndex{1}\)
Which of the following carbocations are likely to undergo a shift? If a shift is likely, draw the new carbocation that would result.
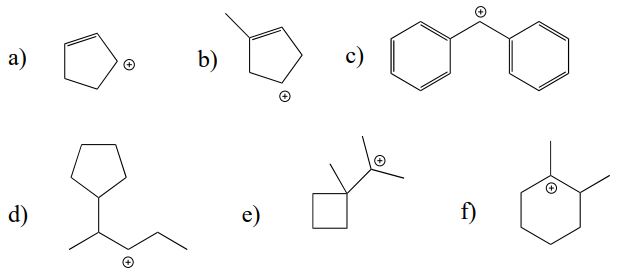
Exercise \(\PageIndex{2}\)
In the (non-biochemcial) reactions below, the major product forms as the result of a hydride or methyl shift from a carbocation intermediate. Predict the structure of the major product for each reaction, disregarding stereochemistry.
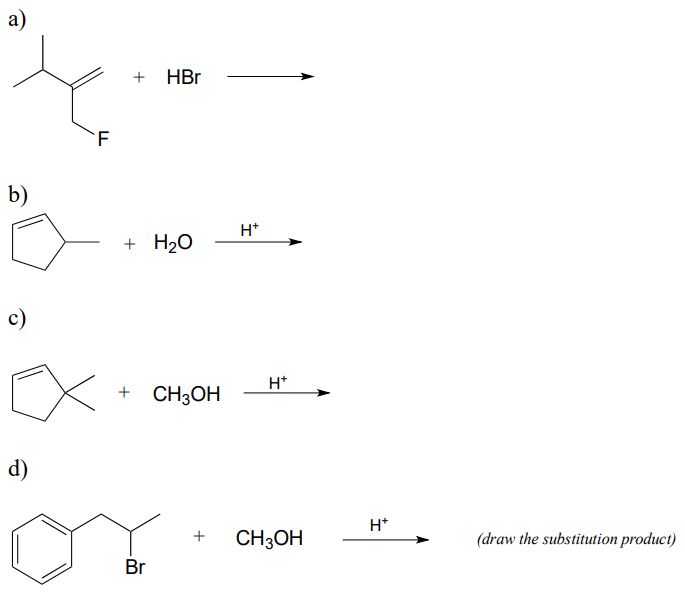
Exercise \(\PageIndex{3}\)
Draw the most abundant product of this laboratory Friedel-Crafts reaction:
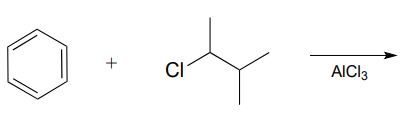
Carbocation rearrangements are involved in many known biochemical reactions. Rearrangements are particularly important in carbocation-intermediate reactions in which isoprenoid molecules cyclize to form complex multi-ring structures. For example, one of the key steps in the biosynthesis of cholesterol is the electrophilic cyclization of oxidosqualene to form a steroid called lanosterol (E.C. 5.4.99.7).

This complex but fascinating reaction has two phases. The first phase is where the actual cyclization takes place, with the formation of four new carbon-carbon bonds and a carbocation intermediate. This phase is a 'cascade' of electrophilic alkene addition steps, beginning with addition of an electrophilic functional group called an 'epoxide'.
The epoxide functional group - composed of a three membered ring with two carbons and an oxygen - is relatively rare in biomolecules and biochemical reactions, and for this reason it is not discussed in detail in this book. However, epoxides are an important and versatile intermediate in laboratory organic synthesis, so you will learn much more about how they are made and how they react if you take a course in chemical synthesis. For now, it is sufficient to recognize that the carbon atoms of an epoxide are potent electrophiles, due to both the carbon-oxygen bond dipoles and the inherent strain of the three membered ring.
The second phase involves a series of hydride and methyl shifts culminating in a deprotonation. In the exercise below, you will have the opportunity to work through the entire cyclase reaction mechanism. In section 15.7, we will take a look at how the epoxide group of oxidosqualene is formed. Trends Pharm. Sci. 2005, 26, 335; J. Phys Chem B., 2012, 116, 13857.
Exercise \(\PageIndex{4}\)
- The figure below outlines the first, cyclizing phase of the reaction that converts oxidosqualene to lanosterol. However, the diagram is missing electron movement arrows, and intermediates 1-4 are all missing formal charges - fill these in.
First phase (ring formation):
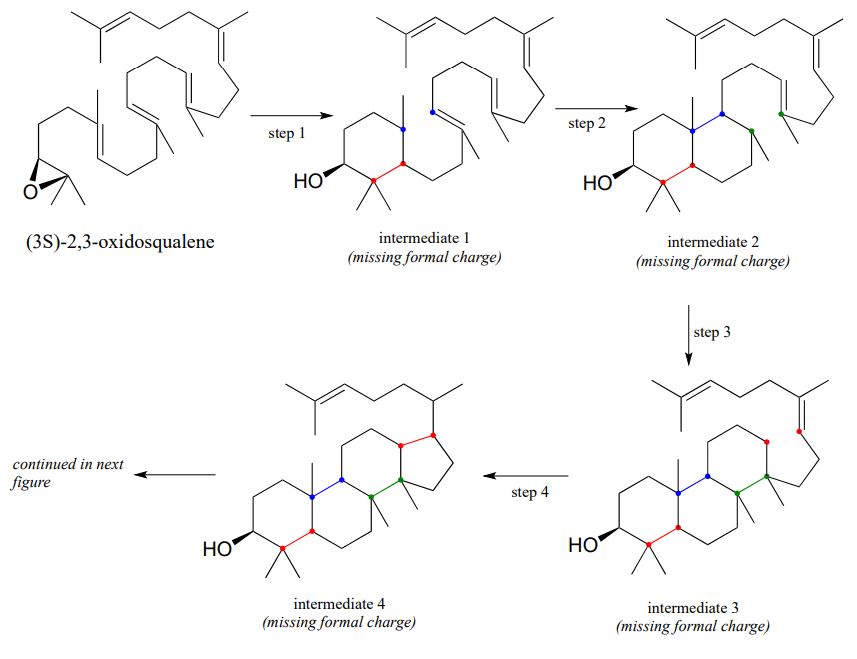
- Next comes the 'shifting' phase of the reaction. Once again, supply the missing mechanistic arrows.
Second phase: rearrangement and deprotonation
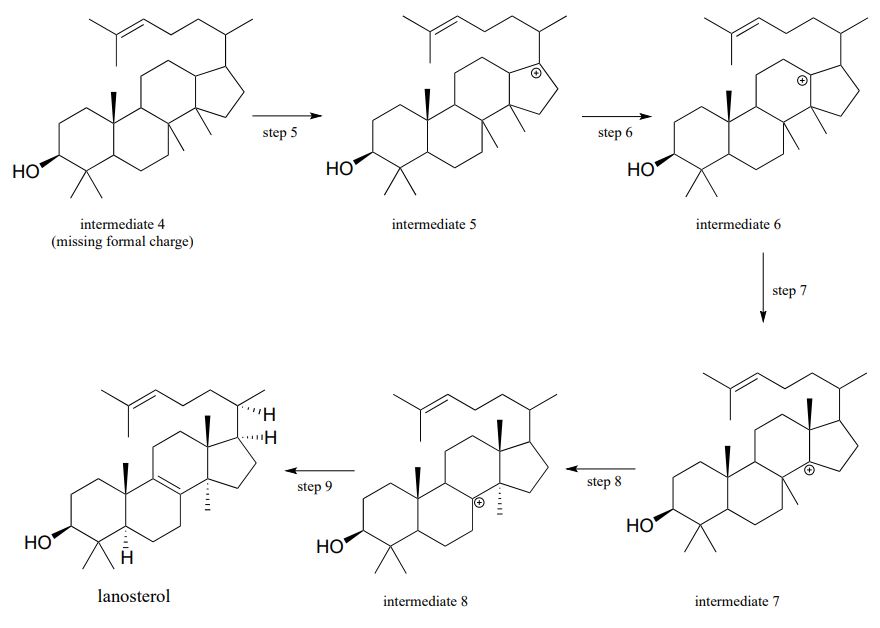
- Look at the first and last steps of the entire process: overall, would you describe this as an electrophilic addition or substitution?
The oxidosqualene cyclization reaction and others like it are truly remarkable examples of the exquisite control exerted by enzymes over the course of a chemical reaction. Consider: an open-chain starting molecule is converted, by a single enzyme, into a complex multiple fused-ring structure with seven chiral centers. Oxidosqualene could potentially cyclize in many different ways, resulting in a great variety of different products. In order for the enzyme to catalyze the formation of a single product with the correct connectivity and stereochemistry, the enzyme must be able to maintain precise control of the conformation of the starting compound and all reactive intermediates in the active site, while also excluding water molecules which could attack at any of the positively charged carbons.
Contributors
Organic Chemistry With a Biological Emphasis by Tim Soderberg (University of Minnesota, Morris)