
Chapter 18: Organic Synthesis
- Page ID
- 23619
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\dsum}{\displaystyle\sum\limits} \)
\( \newcommand{\dint}{\displaystyle\int\limits} \)
\( \newcommand{\dlim}{\displaystyle\lim\limits} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\(\newcommand{\longvect}{\overrightarrow}\)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)‘There could be ART in Organic Synthesis’ declared the inimitable monarch of organic synthesis, Professor R.B. Woodward. His school unveiled several elegant approaches covering a variety of complex structures and broke new grounds to define the art of organic synthesis. ‘If organic synthesis is a branch of science, what is the LOGIC of organic synthesis?’ marveled several others. The development of the concept of logical approaches towards synthesis has been evolving over the past several decades. A few stalwarts focused their attention on this theme and attempted to evolve a pattern to define this logic. There is no doubt that all of us who dabble with synthesis contribute our small bit in the magnificent direction. A few names stand out in our minds for their outstanding contributions. Notable contributions came from the schools of J.A. Marshal, E.J. Wenkert, G. Stock, S Hanessian, E.E. van Tamalen, S. Masamune, R.B. Woodward, E.J. Corey and several others. More focused on this theme were the contributions from the school of E.J. Corey.
The period 1960 – 1990 witnessed the evolution of this thought and the concept bloomed into a full-fledged topic that now merits a separate space in college curriculum. Earlier developments focused on the idea of ANTITHETIC APPROACHES and perfected the art of DISCONNECTION via RETROSYNTHESIS. This led to logical approaches for the construction of SYNTHETIC TREES that summarized various possible approaches for the proposed Target structure. All disconnections may not lead to good routes for synthesis. Once the synthetic tree was constructed, the individual branches were analyzed critically. The reactions involved were looked into, to study their feasibility in the laboratory, their mechanistic pathways were analyzed to understand the conformational and stereochemical implications on the outcome of each step involved and the time / cost factors of the proposed routes were also estimated. The possible areas of pitfall were identified and the literature was critically scanned to make sure that the steps contemplated were already known or feasible on the basis of known chemistry. In some cases, model compounds were first constructed to study the feasibility of the particular reaction, before embarking on the synthesis of the complex molecular architecture. Thus a long process of logical planning is now put in place before the start of the actual synthetic project. In spite of all these careful and lengthy preparations, an experienced chemist is still weary of the Damocles Sword of synthesis viz., the likely failure of a critical step in the proposed route(s), resulting in total failure of the entire project. All achievements are 10% inspiration and 90% perspiration. For these brave molecular engineers, sometimes also called chemists, these long-drawn programs and possible perils of failures are still worth, for the perspiration is enough reward.
A sound knowledge of mechanistic organic chemistry, detailed information on the art and science of functional group transformations, bond formation and cleavage reactions, mastery over separation and purification techniques and a sound knowledge of spectroscopic analysis are all essential basics for the synthesis of molecules. A synthetic chemist should also be aware of developments in synthetic strategies generated over the years for different groups of compounds, which include Rules and guidelines governing synthesis. Since organic chemistry has a strong impact on the development of other sister disciplines like pharmacy, biochemistry and material science, an ability to understand one or more of these areas and interact with them using their terminologies is also an added virtue for a synthetic chemist. With achievements from synthesis of strained molecules (once considered difficult (if not impossible) to synthesize, to the synthesis of complex, highly functionalized and unstable molecules, an organic chemist could now confidently say that he could synthesize any molecule that is theoretically feasible. This is the current status of the power of organic synthesis. Based on the task assigned to the chemist, he would select a Target molecule for investigation and devise suitable routes for synthesis.
Protection and Deprotection Strategies in Organic syntheses
For the manipulation of functional groups and formation of new covalent bonds we make use of a large number of Reagents and Name Reactions. In complex organic syntheses, the starting materials and intermediates in the synthetic scheme often have more than one reactive functional group. A few such multifunctional building blocks are shown below to illustrate this point (Fig 4.1.1 ). While working on such complex
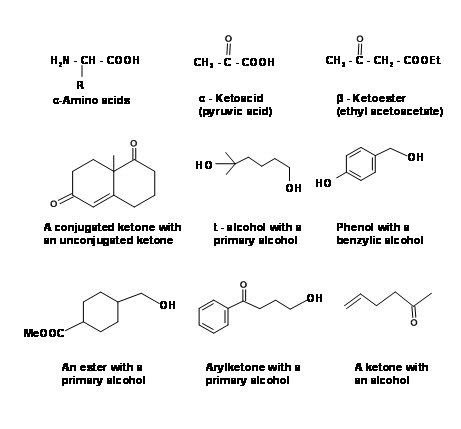
molecules, it is often necessary to protect some groups to enable selective working at the desired locations only. Organic chemists have heavily relied on such protection / deprotection strategies and have diligently developed protecting (masking) and deprotection (unmasking) protocols. We would discuss some of the important protecting groups in this chapter.
Before proceed further, it must be emphasized here that this protocol should be applied only after alternate options have been critically analyzed. This is because protection / deprotection strategy involves an increase of at least two more critical steps, adding to the length of the synthesis and consequent drop in overall yields of the desired compound. In large-scale reactions, this leads to a huge impact to the Atom Economy and pollution cost of the synthetic process. All this translates into an increase in the overall cost of the final drug molecule.
Protection Strategies
Group / Site Selective Reagent: Protection / deprotection is not always required whenever you see a multiplicity of functional groups. You could solve the selectivity issue by using site selective reactions / reagents. By choosing an appropriate selective reagent to suit the scheme on hand, you could selectively attack only one of the reactive sites. Consider an olefinic ketone (Fig 4.1.2). Sodium borohydride reduction in methanol as solvent could selectively reduce the keto- group to a secondary alcohol
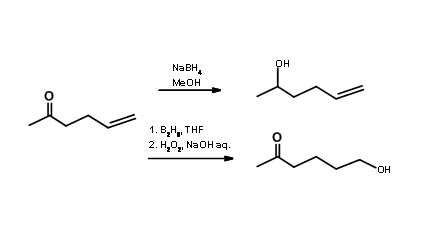
leaving the olefin undisturbed. On the other hand, diborane reagent in THF as solvent would be a reagent of choice when the selective reduction at the olefin moiety is desired. Diborane reduction of an olefin is several times faster than reduction of ketones. The oxidative cleavage of borane product is also selective. Thus, you can avoid the protection / deprotection strategy by employing a selective reagent. In C – C bond formation reactions we come across several such site-selective reagents. One such reagent widely used in research is the Wittig reagent. They attack the aldehyde or ketone selectively in the presence of ester, nitrile. olefin etc..
Selective Protection
In the case of a molecule like 4.1.3A (Fig 4.1.3) bearing an olefin and a carboxylic acid, the –COOH group is several times more reactive than the olefin towards diborane reduction.
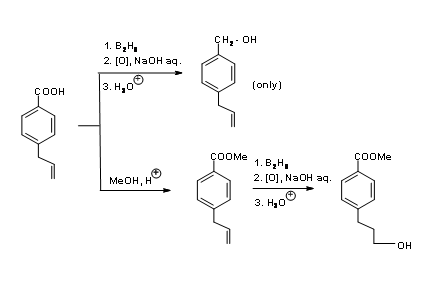
Hydroboration / oxidation reduces the acid to a primary alcohol, leaving the olefin unaffected. On the other hand, if you need a selective reduction of olefin, the acid group has to be processed through a selective protection / deprotection sequence as shown in (Fig 4.1.3)
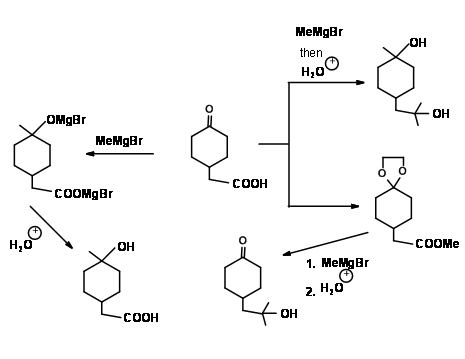
Compound 4.1.4A illustrates several important points in Protection / Deprotection protocol. Both the functional groups could react with a Grignard Reagent. Carboxylic acid group would first react with one mole of the Grignard Reagent to give a carboxylate anion salt. This anion does not react any further with the reagent. When two moles of Grignard Reagent are added to the reaction mixture, the second mole attacks the ketone to give a tertiary alcohol. On aqueous work-up, the acid group is regenerated. Thus, the first mole of the reagent provides a selective transient protection for the –COOH group. Once the acid group is esterified, such selectivity towards this reagent is lost. The reagent attacks at both sites. If reaction is desired only at the ester site, the keto- group should be selectively protected as an acetal. In the next step, the grignard reaction is carried out. Now the reagent has only one group available for reaction. On treatment with acid, the ketal protection in the intermediate compound is also hydrolyzed to regenerated the keto- group.
Orthogonal Protection or Differential Protection
Orthogonal protection is a strategy that allows deprotection of multiple protective groups one at a time, each with a dedicated set of reagents and / reaction conditions without affecting the other. This technique is best illustrated with peptide bond formation and associated deprotection reactions. An amino acid has two functional groups –NH2 and –COOH. When two amino acids (A and B) react under conditions for the peptide bond condensation reaction, a mixture of 4 dipeptides (at least) could be formed as shown below.
\[ \ce{ A + B \rightarrow A-A + A-B + B-A + B-B}\]
If we are interested in only one product A – B, we have to do selective protections and selective deprotections in a proper sequence. Consider the following peptide bond formation reaction.

In order to get only one product A – B, we should protect the N – terminal of ‘A’ and C – terminal of ‘B’. Let us look closely at two different dipeptide formation schemes. In the following sequence, the C – terminal is protected in two different ways for one amino acid. For the second amino acid, the N – terminal is protected with an acid labile Boc- protection.
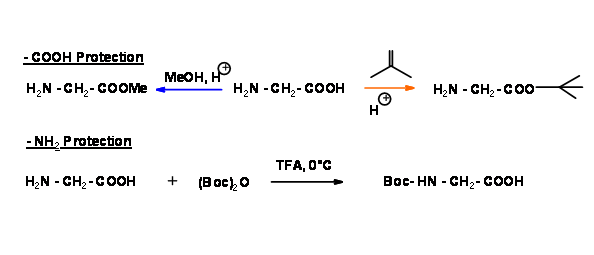
In the next step, the two monoprotected amino acids are coupled as shown below.
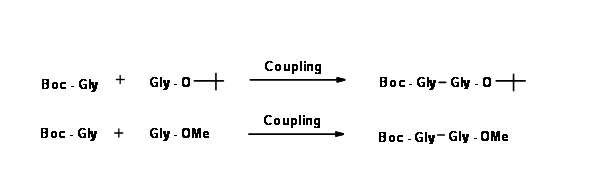
Take a close look at both the products. In the first product, both protections are acid sensitive. If the final product desired is the protection-free dipeptide, this is indeed a short route.
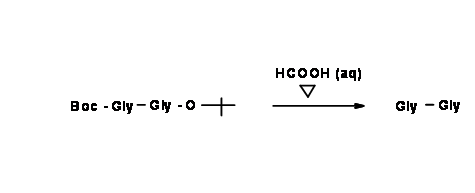
If the desired product is a mono-protected dipeptide, then selective deprotection is the preferred reaction. This is feasible only when we use starting compounds that are differentially protected. This is called Orthogonal Protection.
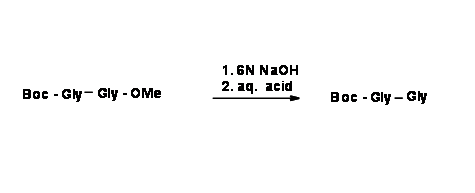
Similar techniques are available for other functional groups as well. Let us now learn more on Protection / Deprotection for some important functional groups.
Protection of R – COOH Group
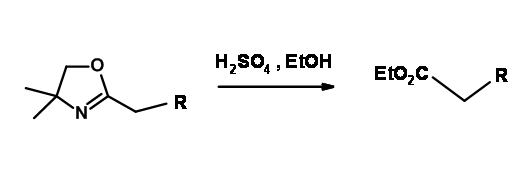
In the introduction, we have seen that carboxylate ion lends protection to an attack of Grignard reagents at this carbonyl carbon. However, this is not sufficient for a vast variety of reagents. Meyer’s 2-oxazolines mask an acid function while activating the α- position for lithiation reaction. The use of this group as protection for –COOH group is rare.
Protection of Aldehydes and Ketones
Since alcohols, aldehydes and ketones are the most frequently manipulated functional groups in organic synthesis, a great deal of work has appeared in their protection / deprotection strategies. In this discussion let us focus on the classes of protecting groups rather than an exhaustive treatment of all the protections.
Acetals
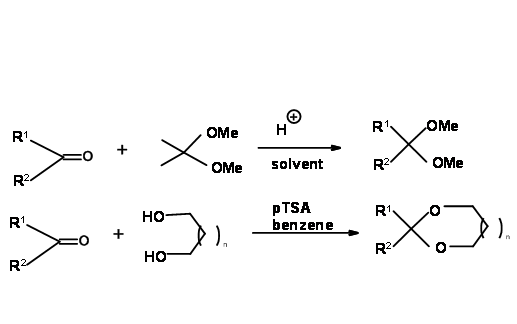
There are two general methods for the introduction of this protection. Transketalation is the method of choice when acetals (ketals) with methanol are desired. Acetone is the by-product, which has to be removed to shift the equilibrium to the right hand side. This is achieved by refluxing with a large excess of the acetonide reagent. Acetone formed is constantly distilled. In the case of cyclic diols, the water formed is continuously removed using a Dean-Stork condenser (Fig 4.1.6).
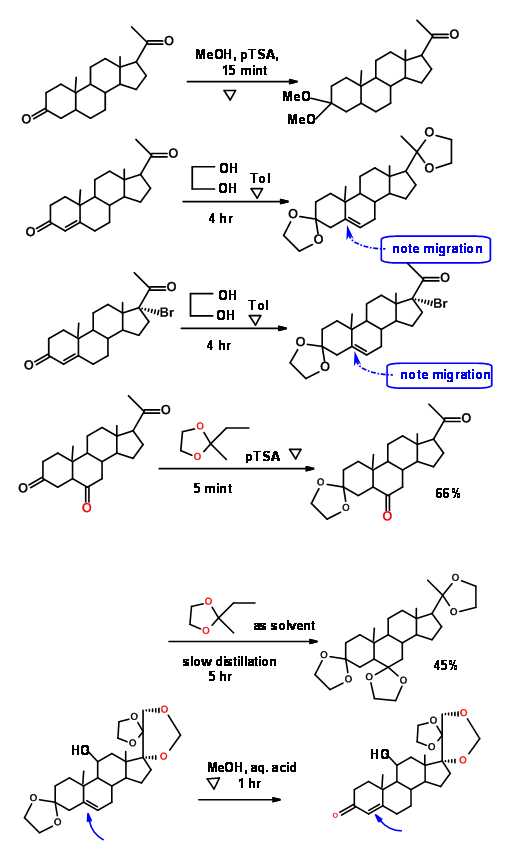
The rate of formation of ketals from ketones and 1,2-ethanediol (ethylene glycol), 1,3-propanediol and 2,2-dimethyl-1,3-propanediol are different. So is the deketalation reaction. This has enabled chemists to selectively work at one center. The following examples from steroid chemistry illustrate these points (Fig 4.1.7).
The demand for Green Chemistry processes has prompted search for new green procedures. Some examples from recent literature are given here (Fig 4.1.8).
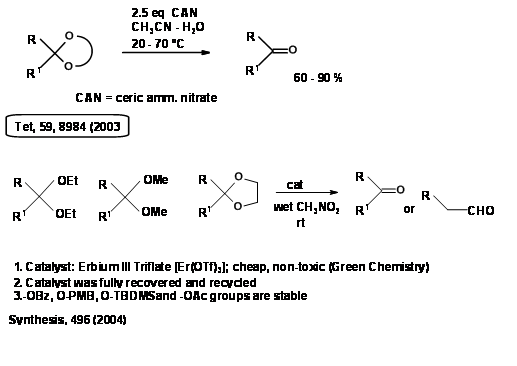
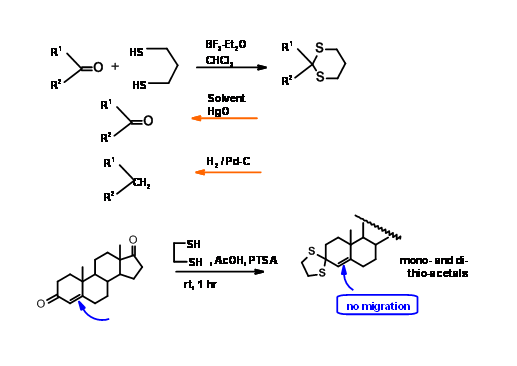
Thioketals
Compared with their oxygen analogues, thioketals markedly differ in their chemistry. The formation as well as deprotection is promoted by suitable Lewis acids. The thioacetals are markedly stable under deketalation conditions, thus paving way for selective operations at two different centers. When conjugated ketones are involved, the ketal formation (as well as deprotection) proceeds with double bond migration. On the other hand, thioketals are formed and deketalated without double bond migration (Fig 4.1.9).
Protection of Amino groups (-NH2 &–NH)
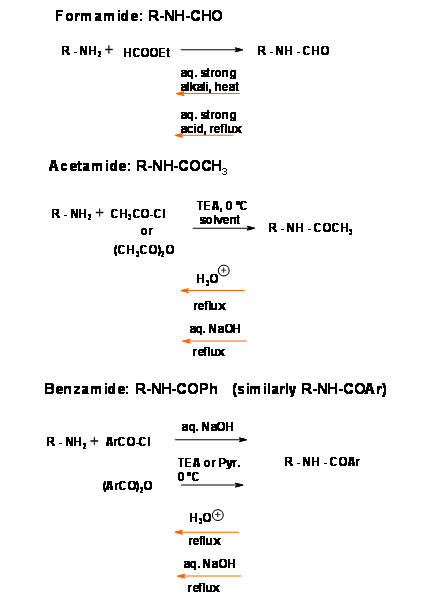
N-Acetyl (N – COCH3), N – Benzoyl (N – COPh) Protections
These are the classical protecting groups for primary and secondary amines. The reagents are cheap and the protocol is simple. Such amides generally need drastic conditions for deprotection, though the yields are generally good (Fig 4.1.10). A standard procedure is refluxing in aqueous alkali or aqueous mineral acid. Due to the drastic conditions, care should be exercised in this procedure to ensure racemi zation is avoided. Amides are generally crystalline solids that are easily purified by crystallization. When the protection is introduced at the early stages of a long synthetic scheme and a very stable protection is desired (as in nucleotide synthesis) an amide is the most preferred protection.
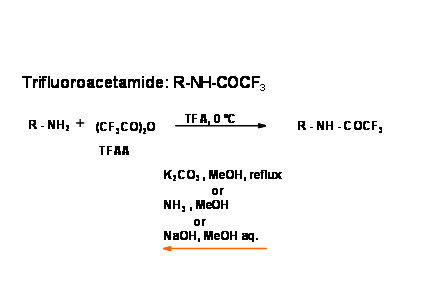
Several more labile amide bonds have been investigated. The amides of trifluoroacetic acid are of special interest. The introduction as well as cleavage is simple and mild {Fig 4.1.11).
A recent report in amide hydrolysis is given below.
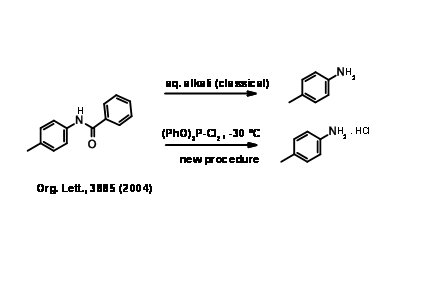
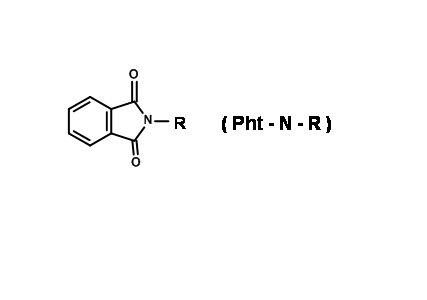
N – Phthaloyl Protection (N – Pht)
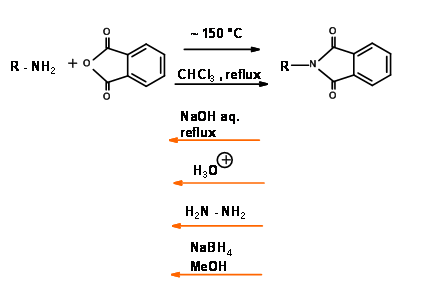
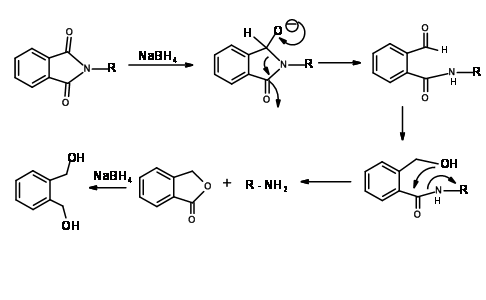
Mechanism for NaBH4 Reduction of N – Pht
N – Carboxylic acid Esters as protective groups
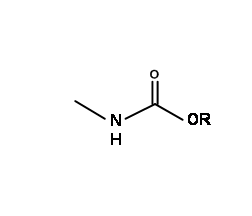
As described above, the amide bonds are very strong. On the other hand, the ester bonds are easily cleaved by mild base conditions. A carboethoxy protection on amine has an amide bond as well as an ester bond. Since N – COOH groups obtained on hydrolysis are very unstable, this protection provides a large family of protective groups for primary and secondary amines.
N – Carboethoxycarbonly (N – COOEt) and Carbobenzyloxycarbonyl (N – COOCH2Ph) (N – Cbz or N – Z) Protections:
These groups are easily introduced using the corresponding chloroformate esters. Anhydrides or mixed anhydrides under mild basic conditions. Both these protections could be removed under prolonged stirring with base at room temperature. Though mild, some racemisation is sometimes observed. The N – Cbz protection has an added advantage in that it could be easily cleaved under hydrogenolysis conditions (Fig 4.1.14). N – Cbz Protection is however stable to acidic conditions. Compare this with –Boc protection discussed below.
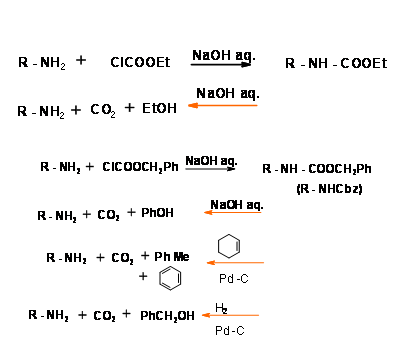
Tert-Butyloxycarbonyl Protection ( N – COOBut, N – Boc)
The Tert-Butyloxycarbonyl Protection could be introduced and removed under very mild acid conditions. This protection is stable to alkali and hydrogenolysis (Fig 4.1.15). Thus, N – Z and N – Boc are complimentary as protective groups.
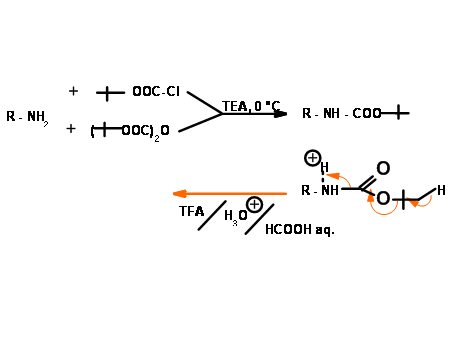
N – Fluoromethyleneoxycarbonyl Protection (Fmoc)
This UV active protecting group is very popular in Solid Phase Peptide Synthesis (SPPS) protocols. Protection as well as deprotection steps proceed under mild conditions in good yields (Fig 4.1.16).
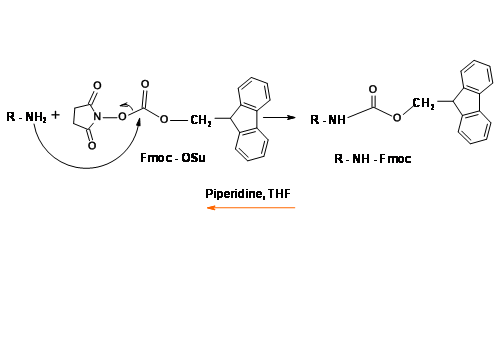
The mechanism for Fmoc deprotection is shown in (Fig 4.1.17)
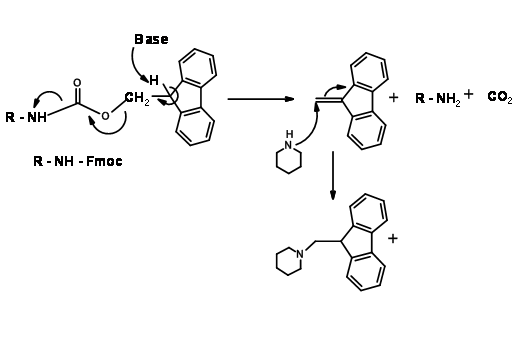
N – Silylation
Silylation is a common protection for active hydrogen on heteroatoms. In the case of N – Si bond, quaternary ammonium fluorides cleave this bond (Fig 4.1.18).
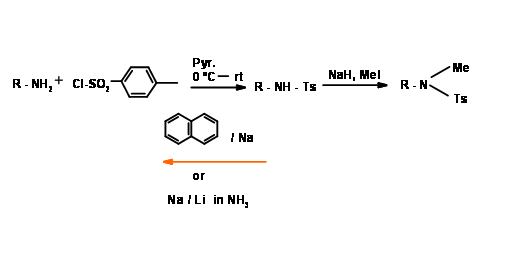
N – Tosylation (N – Tos)
This protection is very stable. N – Tosylation is easily carried out through acid chloride procedure. It is cleaved by solvated electron cleavage reaction. When this group is attached to a primary amine, the –NH group becomes very acidic (Fig 4.1.19).
Protection of – OH Groups
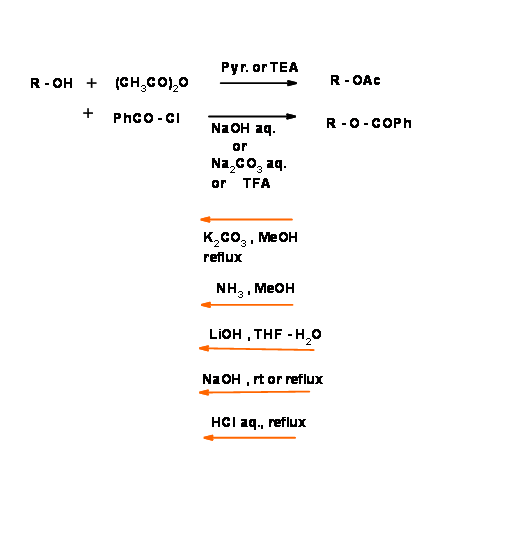
Acetates ( – Ac) and benzoates (– Obz)):
The – OH group protection chemistry has been extensively investigated. The classical protection is the formation of esters of aliphatic and aromatic carboxylic acids. Aromatic esters are comparatively difficult to hydrolyze under mild base condition. This provides an opportunity for selective deprotection protocols (Fig 4.1.20). Note that this protection is sensitive to acid as well as base conditions.
Methyl (– OMe) and Benzyl (R – OBn) Ethers
An ether group is one amongst the most stable functional groups. Hence, this group has been the most favored protecting group. Deprotection was a problem. In the early part of the twentieth century, the only procedure was refluxing with aqueous HI or HBr. In recent years several new procedures have appeared for effective removal under mild conditions. The special feature of the benzyl ethers is that this protection is readily removed under neutral hydrogenolysis conditions (Fig 4.1.21). Substituents like – OMe or – NO2 could be introduced on the benzene ring to modify the reactivity at the protection site.
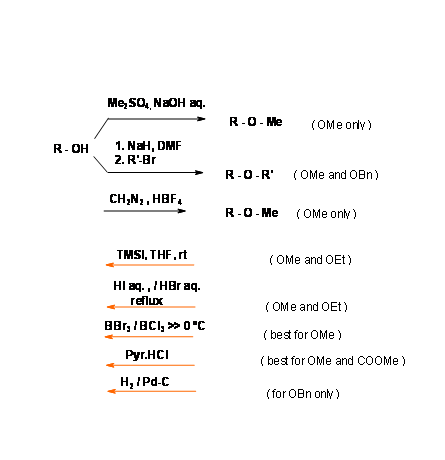
When an olefin could compete at the hydrogenolysis procedure, the following sequence appears to be an alternate procedure (Fig 4.1.22).
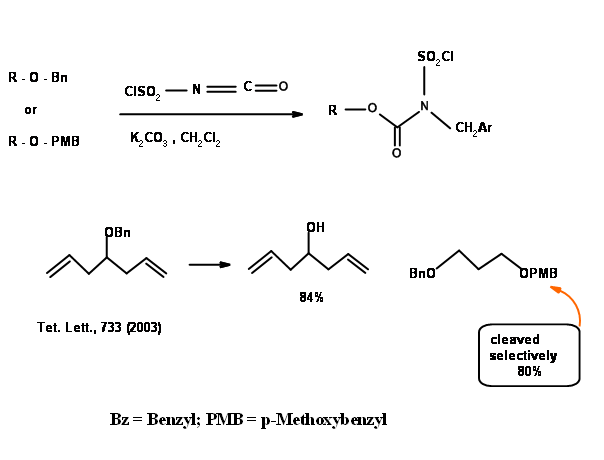
Allyl ether is a recent introduction in – OH protection. The versatility of this protection could be seen in the following examples (Fig 4.1.23).
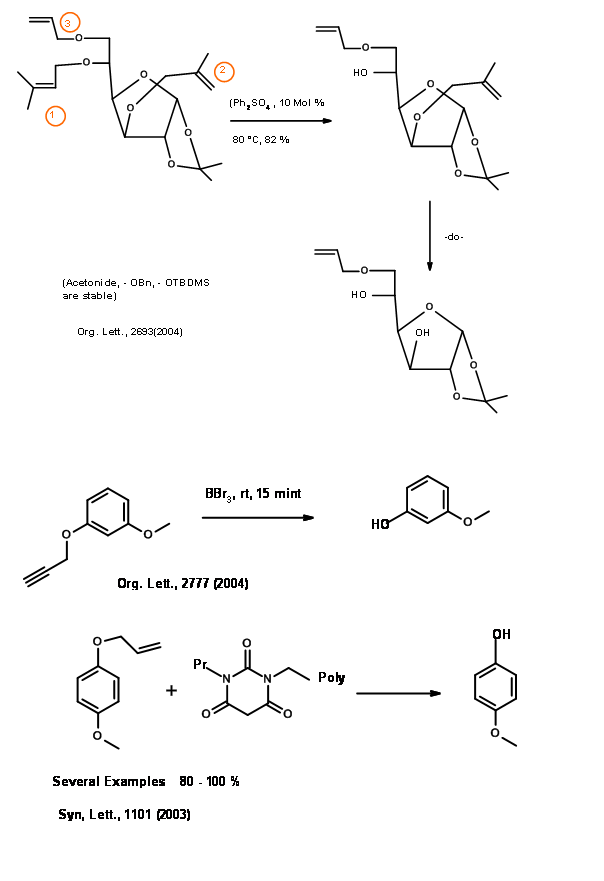
Silyl Ethers (R – OSiR3)
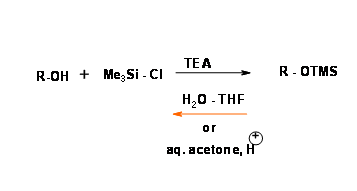
The oxygen – silicon sigma bond is stable to lithium and Grignard reagents, nucleophiles and hydride reagents but very unstable to water and mild aqueous acid and base conditions. A silyl ether of secondary alcohol is less reactive than that of a primary alcohol. The O – trimethylsilyl (O – SiMe3) was first protection of this class. (Fig 4.1.24).
Replacement of methyl group with other alkyl and aryl groups gives a large variety of silyl ether with varying degrees of stability towards hydrolysis (Fig 4.1.25).

The following examples illustrate the selectivity in formation and hydrolysis of this group (Fig 4.1.26).
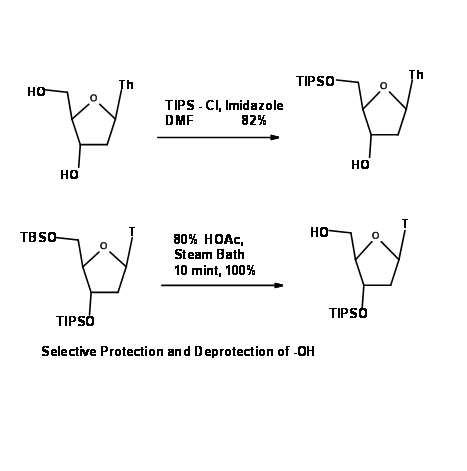
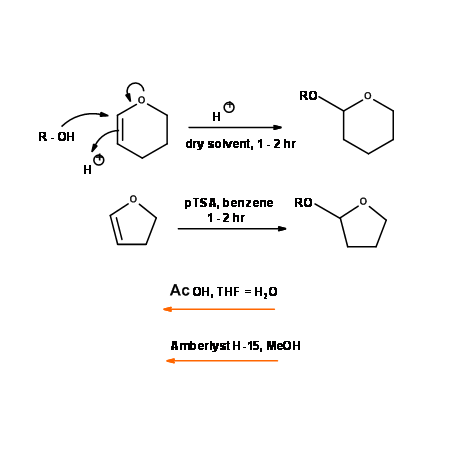
Tetrahydropyranyl ether (– OTHP) and Tetrahydrofuranyl ether (– OTHF)
These protective groups for alcohols are in fact acetals. They are synthesized using the dihydropyran (DHP) and dihydrofuran (DHF) respectively. They behave like acetals in their stability and cleavage (Fig 4.1.27). The rate of formation and cleavage for these two groups differ, which finds application for differential protection of alcohols.
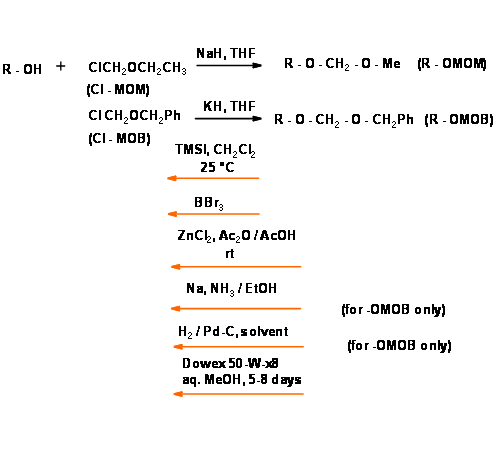
These protective groups found extensive use in synthesis. However, two major drawbacks were soon observed.
- A new stereopoint is generated while introducing this protection. Though it is not relevant from the point of view of the target molecule, in chiral molecules this created diastereomer problems in spectroscopy (NMR and MS) and chromatography.
- These ethers occasionally caused explosions in hydroboration procedures due to peroxide formation. The diastereomer problem was solved by the introduction of O-methyleneoxymethyl ether ( – O – MOM) and O – methyleneoxybenzyl ether (R – O – MOB) (Fig 4.1.28). Several other modifications are now available.
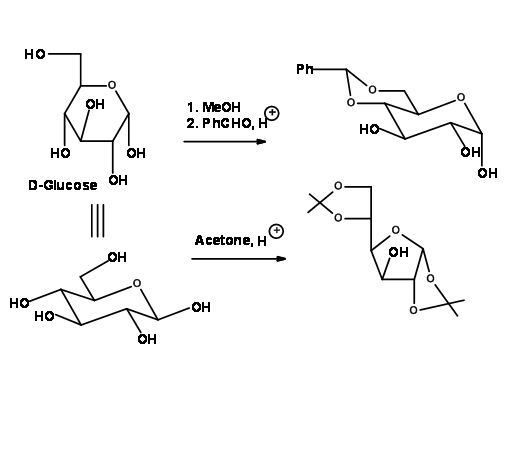
Protection of vic – Diols
On reaction with benzaldehyde or acetone with suitable acid catalyst, vic-diols form cyclic acetals. This in fact is a proof for the existence of vic-diols in the molecule. They are acetal protections and therefore behave as acetals in their chemistry (Fig 4.1.29)
Conclusion
The above discussions are just a glimpse of the vast literature on this topic. When more than one competing functional groups are present in a molecule, it may be necessary to introduce at least one protection and one deprotection step in the synthetic scheme. This adds not only to the length of the synthetic scheme, but also to the cost of the final compound. With growing awareness in Green Chemistry, chemists have been trying to reduce this protocol to a minimum or preferably avoid this altogether. Several protection free syntheses of natural products are known in the literature. We would discuss this topic at the end of this chapter.
Further Reading
- Greene T. W., Protective Groups in Organic Synthesis. Wiley. N. Y., (1980), (1991).
- Smith M. B., Organic Synthesis, McGraw-Hill Inc, N. Y., (1994).
- Djerassi C., Steroid Reactions –An Outline for Organic Chemists, Holden-Day nc. San Francisco )1963).
- Advanced Organic Chemistry: Principles Tools and Logic of Synthesis, R. Balaji Rao, Vishal Publishing Co., Jalandhar, India (2012).
- Amino Acids, Peptides and Proteins in Organic Chemistry, Vol 4, Ed by Andrew B. Hughes (2011) Wiley-VCH; Protection Reactions, V.V. Sureshbabu and N. Narendra page 1 – 97.
4.2 Disconnection of bonds
Having chosen the TARGET molecule for synthesis, the next exercise is to draw out synthetic plans that would summarize all reasonable routes for its synthesis. During the past few decades, chemists have been working on a process called RETROSYNTHESIS. Retrosynthesis could be described as a logical Disconnection at strategic bonds in such a way that the process would progressively lead to easily available starting material(s) through several synthetic plans. Each plan thus evolved, describes a ‘ROUTE’ based on a retrosynthesis. Each disconnection leads to a simplified structure. The logic of such disconnections forms the basis for the retroanalysis of a given target molecule. Natural products have provided chemists with a large variety of structures, having complex functionalities and stereochemistry. This area has provided several challenging targets for development of these concepts. The underlining principle in devising logical approaches for synthetic routes is very much akin to the following simple problem. Let us have a look of the following big block, which is made by assembling several small blocks (Fig 4.2.1). You could easily see that the large block could be broken down in different ways and then reassembled to give the same original block.
Now let us try and extend the same approach for the synthesis of a simple molecule. Let us look into three possible ‘disconnections’ for a cyclohexane ring as shown in Figure 4.2.2.
In the above analysis we have attempted to develop three ways of disconnecting the six membered ring. Have we thus created three pathways for the synthesis of cyclohexane ring? Do such disconnections make chemical sense? The background of an organic chemist should enable him to read the process as a chemical reaction in the reverse (or ‘retro-‘) direction. The dots in the above structures could represent a carbonium ion, a carbanion, a free radical or a more complex reaction (such as a pericyclic reaction or a rearrangement). Applying such chemical thinking could open up several plausible reactions. Let us look into path b, which resulted from cleavage of one sigma bond. An anionic cyclisation route alone exposes several candidates as suitable intermediates for the formation of this linkage. The above analysis describes only three paths out of the large number of alternate cleavage routes that are available. An extended analysis shown below indicates more such possibilities (Fig 4.2.3). Each such intermediate could be subjected to further disconnection process and the process continued until we reach a reasonably small, easily available starting materials. Thus, a complete ‘SYNTHETIC TREE’ could be constructed that would summarize all possible routes for the given target molecule.
4.3 Efficiency of a route
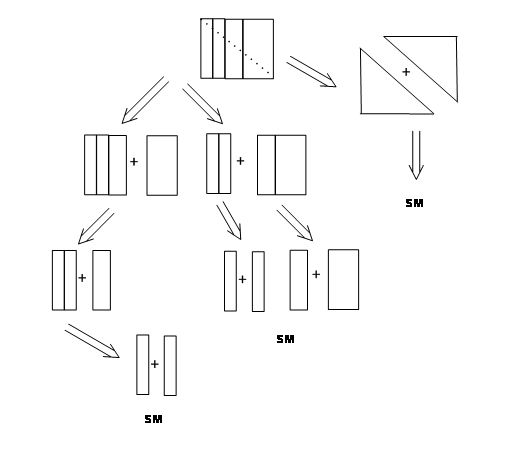
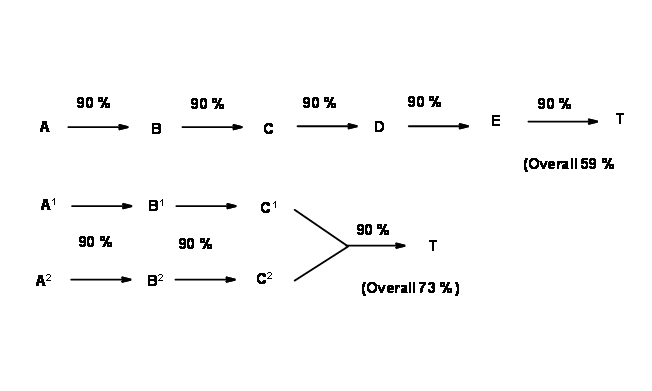
A route is said to be efficient when the ‘overall yield’ of the total process is the best amongst all routes investigated. This would depend not only on the number of steps involved in the synthesis, but also on the type of strategy followed. The strategy could involve a ‘linear syntheses’ involving only consequential steps or a ‘convergent syntheses’ involving fewer consequential steps. Figure 4.3.1 shown below depicts a few patterns that could be recognized in such synthetic trees. When each disconnection process leads to only one feasible intermediate and the process proceeds in this fashion
all the way to one set of starting materials (SM), the process is called a Linear Synthesis. On the other hand, when an intermediate could be disconnected in two or more ways leading to different intermediates, branching occurs in the plan. The processes could be continued all the way to SMs. In such routes different branches of the synthetic pathways converge towards an intermediate. Such schemes are called Convergent Syntheses.
The flow charts shown below (Fig 4.3.2) depicts a hypothetical 5-step synthesis by the above two strategies. Assuming a very good yield (90%) at each step (this is rarely seen in real projects), a linier synthesis gives 59% overall yield, whereas a convergent synthesis gives 73% overall yield for the same number of steps..
4.4 Problem of substituents and stereoisomers
The situation becomes more complex when you consider the possibility of unwanted isomers generated at different steps of the synthesis. The overall yield drops down considerably for the synthesis of the right isomer. Reactions that yield single isomers (Diastereospecific reactions) in good yields are therefore preferred. Some reactions like the Diels Alder Reaction generate several stereopoints (points at which stereoisomers are generated) simultaneously in one step in a highly predictable manner. Such reactions are highly valued in planning synthetic strategies because several desirable structural features are introduced in one step. Where one pure enantiomer is the target, the situation is again complex. A pure compound in the final step could still have 50% unwanted enantiomer, thus leading to a drastic drop in the efficiency of the route. In such cases, it is desirable to separate the optical isomers as early in the route as possible, along the synthetic route. This is the main merit of the Chiron Approach, in which the right starting material is chosen from an easily available, cheap ‘chiral pool’. We would discuss this aspect after we have understood the logic of planning syntheses. Given these parameters, you could now decide on the most efficient route for any given target.
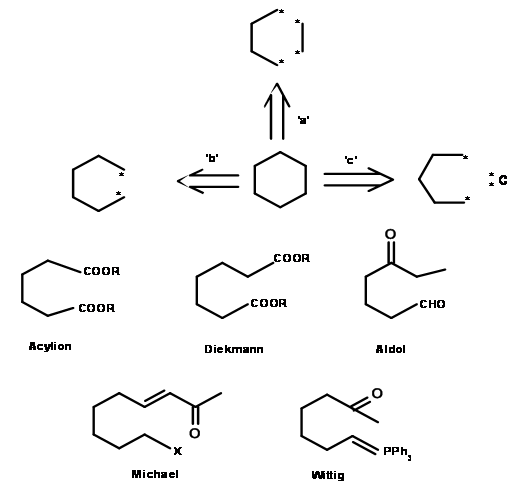
Molecules of interest are often more complex than the plain cyclohexane ring discussed above. They may have substituents and functional groups at specified points and even specific stereochemical points. Construction of a synthetic tree should ideally accommodate all these parameters to give efficient routes. Let us look into a slightly more complex example shown in Figure 4.4.1 . The ketone 4.4.1A is required as an intermediate in a synthesis. Unlike the plain cyclohexane discussed above, the substitution pattern and the keto- group in this molecule impose some restrictions on disconnection processes.
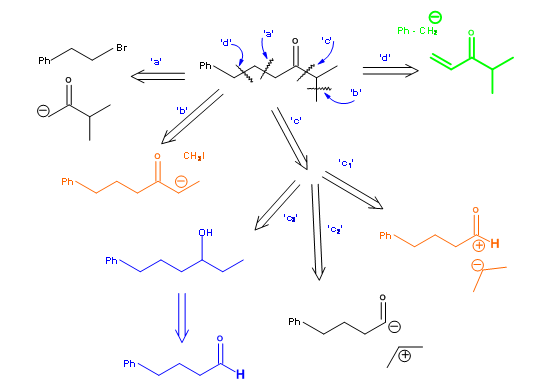
Cleavage a: This route implies attack of an anion of methylisopropylketone on a bromo-component. Cleavage b: This route implies simple regiospecific methylation of a larger ketone that bears all remaining structural elements. Cleavage c: This route implies three different possibilities. Route C-1 envisages an acylonium unit, which could come from an acid halide or an ester. Route C-2 implies an umpolung reaction at the acyl unit. Route C-3 suggests an oxidation of a secondary alcohol, which could be obtained through a Grignard-type reaction. Cleavage d: This implies a Micheal addition.
Each of these routes could be further developed backwards to complete the synthetic tree. These are just a few plausible routes to illustrate an important point that the details on the structure would restrict the possible cleavages to some strategic points. Notable contributions towards planning organic syntheses came from E.J. Corey’s school. These developments have been compiles by Corey in a book by the title LOgIC OF CHEMICAL SYNTHESIS. These and several related presentations on this topic should be taken as guidelines. They are devised after analyzing most of the known approaches published in the literature and identifying a pattern in the logic. They need not restrict the scope for new possibilities. Some of the important strategies are outlined below.
4.5 Preliminary scan
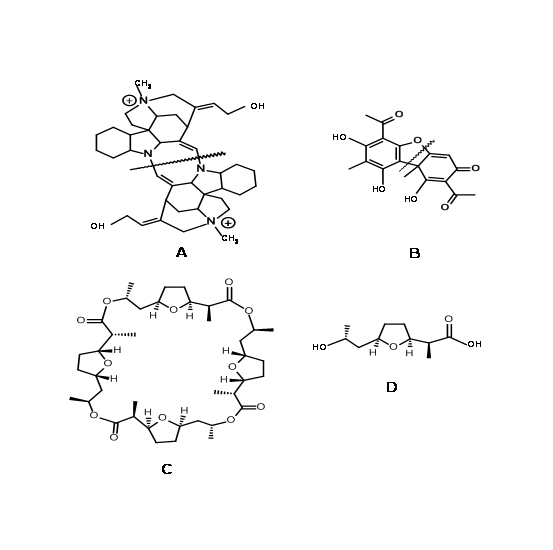
When a synthetic chemist looks at the given Target, he should first ponder on some preliminary steps to simplify the problem on hand. Is the molecule polymeric? See whether the whole molecule could be split into monomeric units, which could be coupled by a known reaction. This is easily seen in the case of peptides, nucleotides and organic polymers. This could also be true to other natural products. In molecules like C-Toxiferin 1 (4.5.1A) (Fig 4.5.1), the point of dimerisation is obvious. In several other cases, a deeper insight is required to identify the monomeric units, as is the case with Usnic acid (4.5.1B). In the case of the macrolide antibiotic Nonactin (4.5.1C), this strategy reduces the possibilities to the synthesis of a monomeric unit (4.5.1D). The overall structure has S4 symmetry and is achiral even though assembled from chiral precursors. Both (+)-nonactic acid and (−)-nonactic acid (4.5.1D) are needed to construct the macrocycle and they are joined head-to-tail in an alternating (+)-(−)-(+)-(−) pattern. (see J. Am. Chem. Soc., 131, 17155 (2009) and references cited therein).
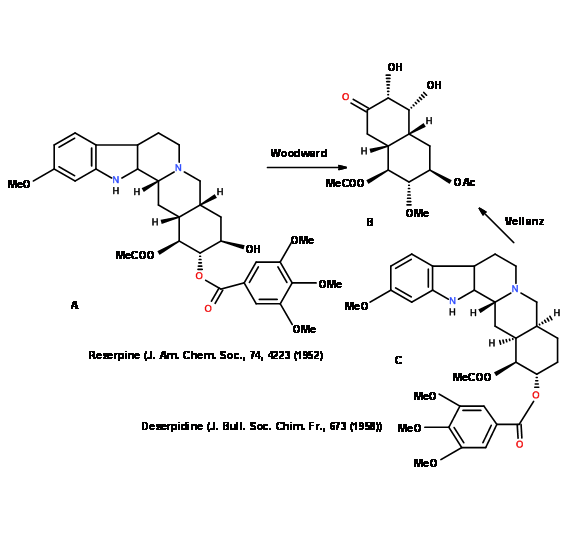
Is a part of the structure already solved? Critical study of the literature may often reveal that the same molecule or a closely related one has been solved. R.B. Woodward synthesized (4.5.2C) as a key intermediate in an elegant synthesis of Reserpine (4.5.2A). The same intermediate compound (4.5.2C) became the key starting compound for Velluz et.al., in the synthesis of Deserpidine (4.5.2B) (Fig 4.5.2).
Such strategies reduce the time taken for the synthesis of new drug candidates. These strategies are often used in natural product chemistry and drug chemistry. Once the preliminary scan is complete, the target molecule could be disconnected at Strategic Bonds.
4.6 Strategic Bonds, Retrons and Transforms
STRATEGIC BONDS are the bonds that are cleaved to arrive at suitable Starting Materials (SM) or SYNTHONS. For the purpose of bond disconnection, Corey has suggested that the structure could be classified according to the sub-structures generated by known chemical reactions. He called the sub-structures RETRONS and the chemical transformations that generate these Retrons were called TRANSFORMS. A short list of Transforms and Retrons are given below (TABLE 4.6.1). Note that when Transforms generate Retrons, the product may have new STEREOPOINTS (stereochemical details) generated that may need critical appraisal.
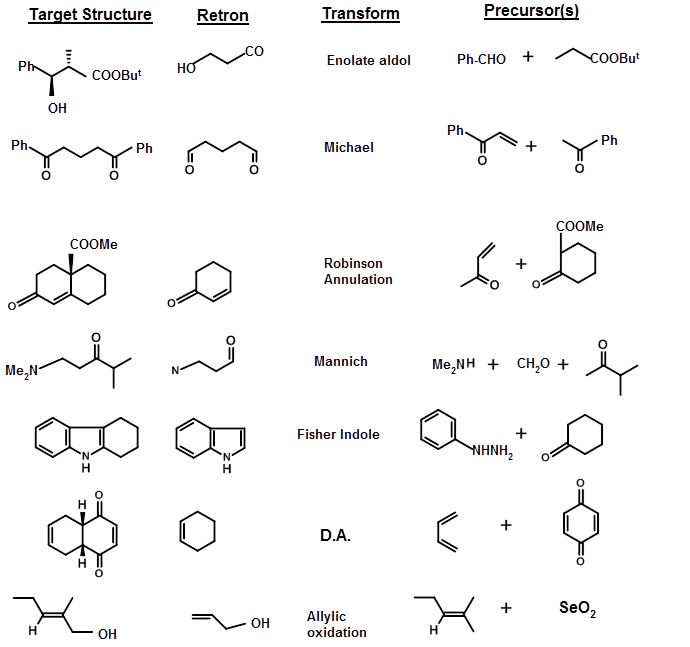
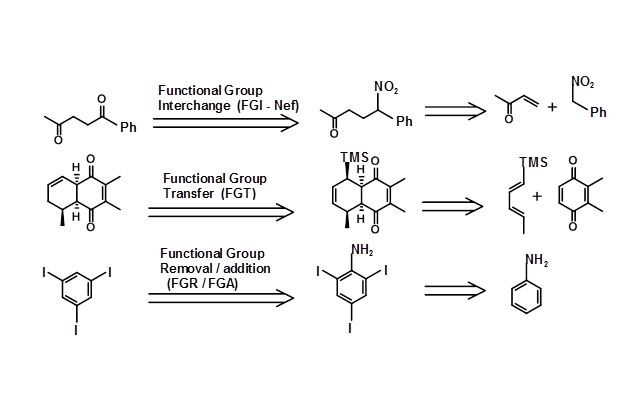
The structure of the target could be such that the Retron and the corresponding Transforms could be easily visualized and directly applied. In some cases, the Transforms or the Retrons may not be obvious. In several syntheses, transformations do not simplify the molecule, but they facilitate the process of synthesis. For example, a keto- group could be generated through modification of a -CH-NO2 unit through a Nef reaction. This generates a new set of Retron / transforms pair. A few such transforms are listed below, along with the nomenclature suggested by Corey (Fig 4.6.2).
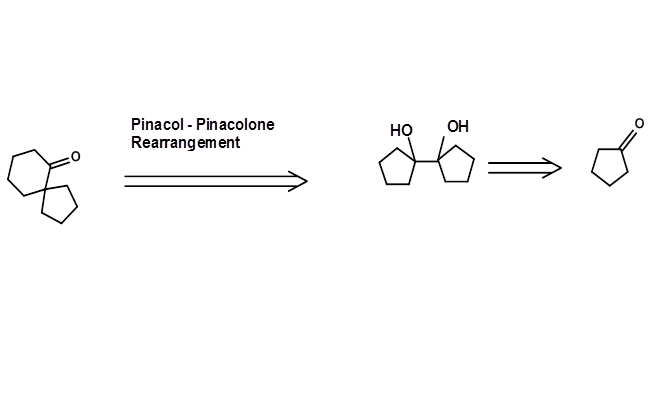
A Rearrangement Reaction could be a powerful method for generating suitable new sub-structures. In the following example, a suitable Pinacol Retron, needed for the rearrangement is obtained through an acyloin transform (Fig 4.6.3). Such rearrangement Retrons are often not obvious to inexperienced eyes.
Some transforms may be necessary to protect (acetals for ketones), modify (reduction of a ketone to alcohol to avoid an Aldol condensation during a Claisen condensation) or transpose a structural element such as a stereopoint (e.g. SN2 inversion, epimerization etc.,) or shifting a functional group. Such transforms do not simplify the given structural unit. At times, activation at specific points on the structure may be introduced to bring about a C-C bond formation and later the extra group may be removed. For example, consider the following retrosynthesis in which an extra ester group has been introduced to facilitate a Dieckmann Retron. In complex targets, combinations of such strategies could prove to be a very productive strategy in planning retrosynthesis. Witness the chemical modification strategy shown below for an efficient stereospecific synthesis of a trisubstituted olefin (Fig 4.6.4)
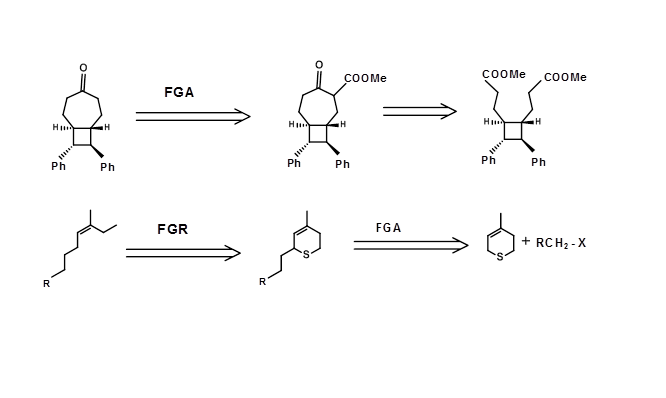
Figure 4.6.4 Examples for FGA / FGR strategies for complex targets
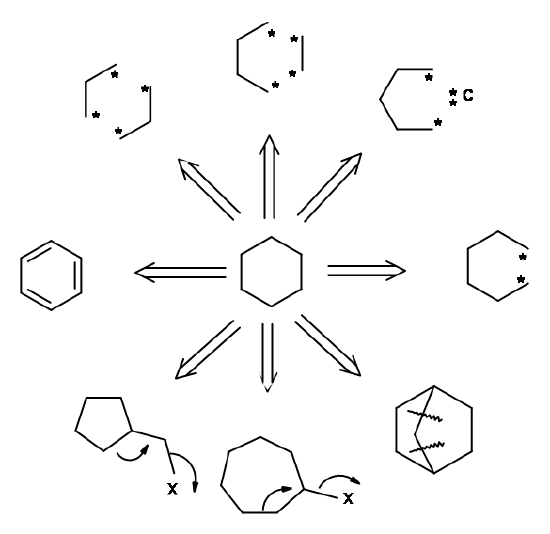
Amongst the molecular architectures, the bridged-rings pose a complex challenge in Structure-Based disconnection procedures. Corey has suggested guidelines for efficient disconnections of strategic bonds.
A bond cleavage for retrosynthesis should lead to simplified structures, preferably bearing five- or six-membered rings. The medium and large rings are difficult to synthesize stereospecifically. Amongst the common rings, a six-membered ring is easily approached and manipulated to large and small rings. Simultaneous cleavage of two bonds, suggesting cycloaddition – retrons are often more efficient. Some cleavages of strategic bonds are shown in Figure 4.6.5, suggesting good and poor cleavage strategies based on this approach. However, these guidelines are not restrictive.
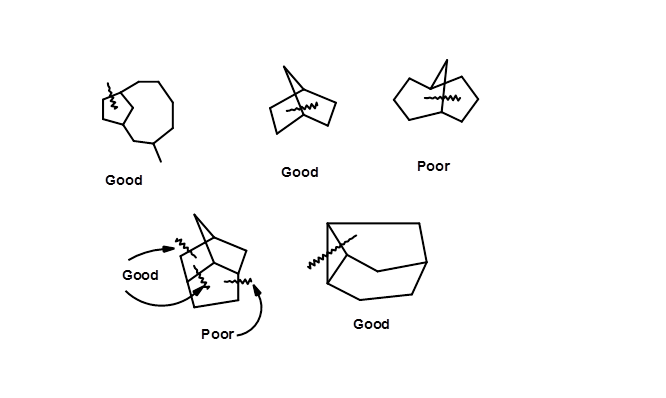
Identifying Retron – Transform sets in a given target molecule is therefore a critical component in retrosynthesis. Such an approach could often generate several synthetic routes. The merit of this approach is that starting materials do not prejudice this logic. Retrosyntheses thus developed could throws open several routes that need further critical scrutiny on the basis of known facts.
Identification of Retrons / Transforms sets provided the prerequisite for computer assisted programs designed for generating retrosynthetic routes. A list of Retrons and the corresponding transforms were interlinked and the data was stored in the computer. All known reactions were thus analyzed for their Retron / Transform characteristics and documented. The appropriate literature citations were also documented and linked. Based on these inputs, computer programs were designed to generate retrosynthetic routes for any given structure. Several such programs are now available in the market to help chemists generate synthetic strategies. Given any structure, these programs generate several routes. Once the scientist identifies the specific routes of interest for further analysis, the program generates detailed synthetic steps, reagents required and the appropriate citations. In spite of such powerful artificial intelligence, the intelligence and intuitive genius of a chemist is still capable of generating a new strategy, not yet programmed. Again, human intelligence is still a critical input for the analysis the routes generated using a computer. Based on the experience of the chemists’ team, their projected aim of the project and facilities available, the routes are further screened.
4.7 Elaboration of the concepts
Short lists of syntheses that exemplify retroanalysis strategies devised through powerful transforms are given below. Several syntheses from natural product chemistry are later discussed in this chapter, which further illustrates these points.
Retrosynthesis based on Diels-Alder Transform; (E.J. Corey et.al., J.A.C.S. (1972), 94, 2549). Fumagillol (4.7.1A) presents 4 stereocentres and sensitive functionalities.
Simplification of the functional groups first exposed a vic-diol. This site could come from an olefin D. Further retroanlysis led to a structurally simplified target sequence B to F. A cyclohexene ring system is suitable for a powerful DA Transform. This step generated two stereocentres in one reaction and also an olefin in the correct position for hydroxylation. The key intermediate C could also be generated through a functional group transform leading to G. This provided scope for a new set of starting materials using another DA Transform. The Retrosynthetic analysis and the actual synthesis are shown in Figure 4.7.1.
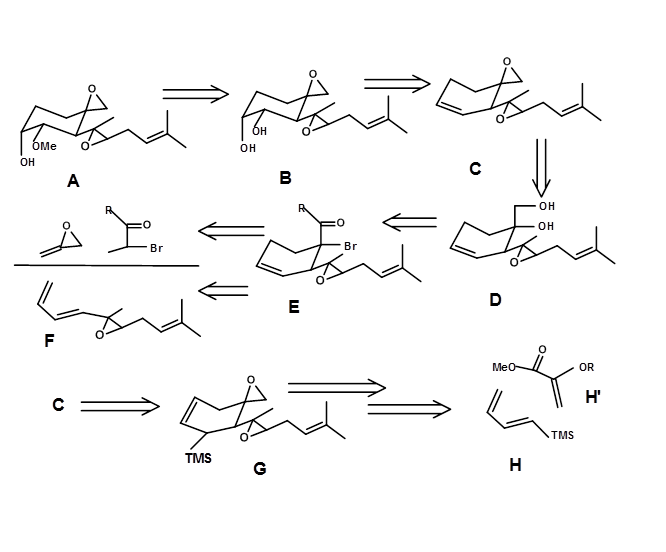
Synthetic protocol reported by Corey is outlined in Figure 4.7.2
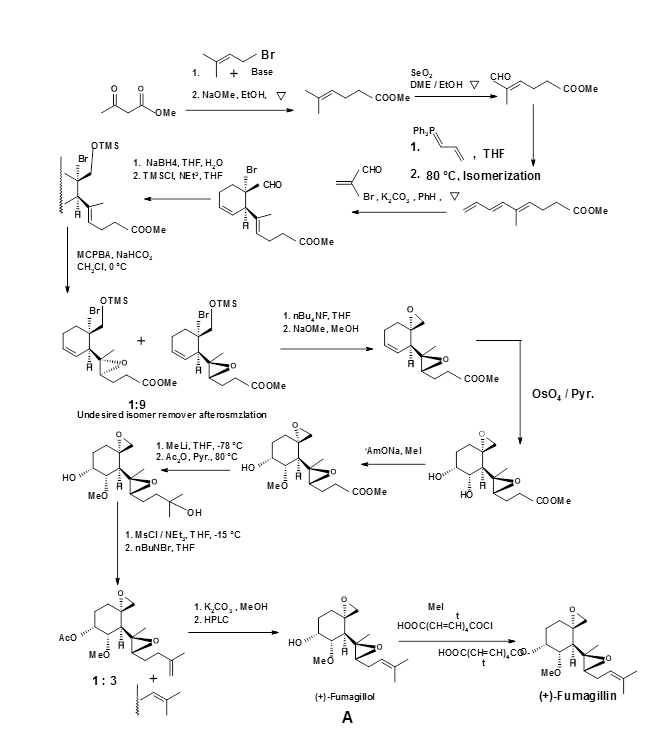
For the synthesis of Estrone, an interesting DA Transform strategy was devised by Kametani et.al.. The retrosynthetic strategy is depicted in Fig 4.7.3. The required diene precursor was generated via cyclo-reversion reaction of a cyclobutene unit (T. Kametani et.al., Tetrahedron, (1981), 37, 3).
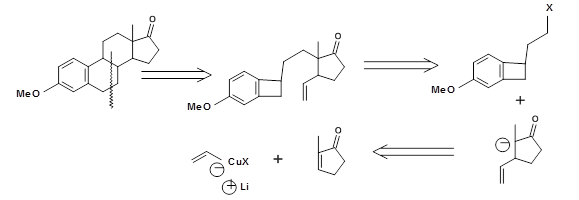
The crucial stereospecific trisubstituted olefins on Squalene (4.6.4B) were synthesized using a Claisen Retron 4.7.4A (Fig 4.7.4). Note the double Claisen approach in this strategy.
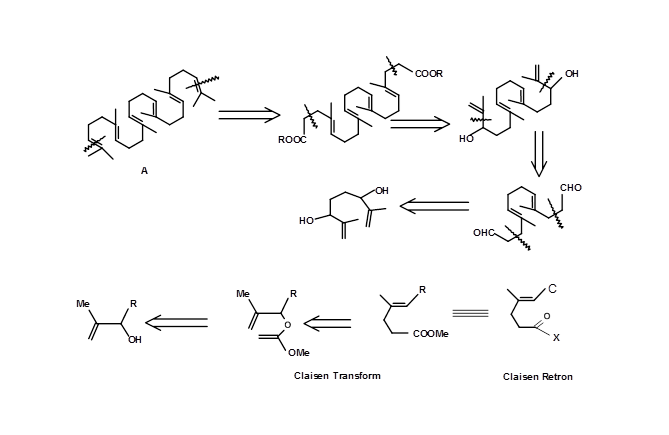
The biogenetic-type cyclisation of olefins provides scope for application of Mechanistic Transform or transforms based on mechanistic considerations. A cleaver introduction of a chiral centre provided an efficient route for generating several enantiopure chiral centres in one step using this strategy (Fig 4.7.5).
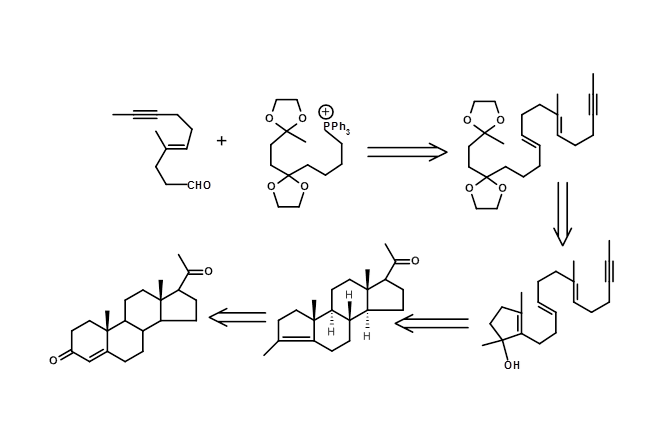
4.8 The problem of enantiomers
In these lengthy discussions above, we learnt about disconnection approaches. We said that stereocentres could introduce special challenges in planning efficient synthetic routes. Let us look at the molecule Biotin to understand disconnection strategies and problem of stereocentres.
Baker established the structure of Biotin in 1947 through an unambiguous synthesis of the molecule. A retroanalysis of the synthetic scheme is as shown below (Fig 4.8.1).
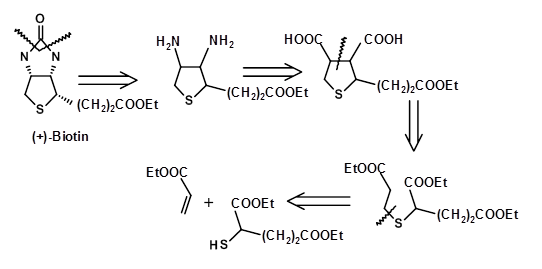
The SM chosen and the synthetic approach clearly established the atom connectivities and the overall structure of the compound. However, the route made not attempt to synthesize one pure isomer because the actual stereochemistry was not established at that time. The route yielded all the eight stereoisomers (3 asymmetric centers). These isomers were carefully separated. In 1952 the biologically active isomer was identified as the all cis- enantiomer (+)-Biotoin. At this stage, several groups reported the stereospecific synthesis of the all cis- isomer exclusively (Fig 4.8.2). The following retroanalysis depicts three such attempts. Note that these efforts were directed towards the synthesis of the racemate and not the pure (+)- isomer of Biotin.
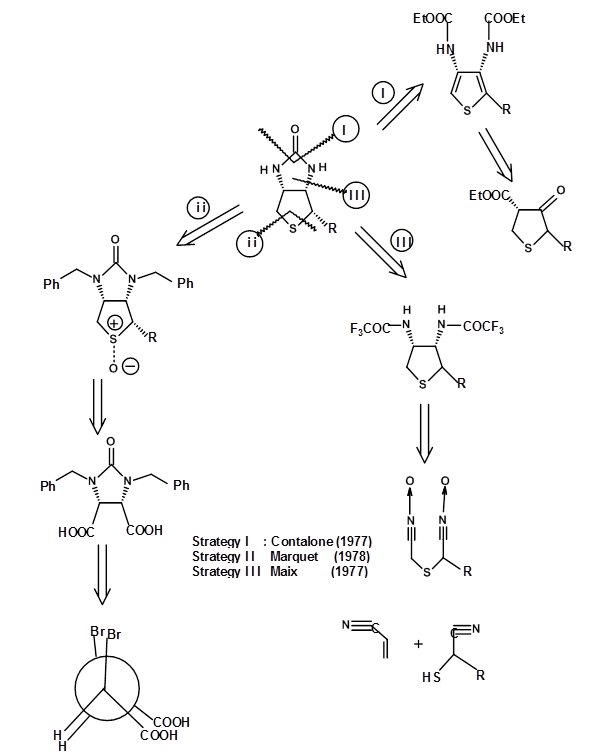
These approaches solved the problem of diastereomeric purity. But they still left a mixture of two unresolved enentiomer viz., (±)-Biotin. To obtain a pure enantiomer in excellent yields, you have to resolve the racemic mixtures at appropriate stages. Alternately one could resort to asymmetric synthesis at all crucial stages. A still better approach would be to start from a chiral SM, which has most of the stereocentres in the correct fashion. This elegant approach is called the Chiron approach. When carefully executed, such procedures yield very pure enantiomer as the final product. Two such approaches for (+)-Biotin is shown below. In the first approach a chiral amino acid cysteine is chosen because it has one key asymmetric center, the sulphur moiety and a carboxylic acid in the correct positions (Fig 4.8.3). In this example the choice of the SM is quite obvious. Note the cleaver introduction of the second nitrogen and the cyclisation step leading to the formation of the tetrahydrothiophene ring. Also note that the yield of Cram vs anti-Cram (chelation) products could be influenced by a choice of the reagent. These kinds of insights come only through a thorough knowledge of this particular reaction.
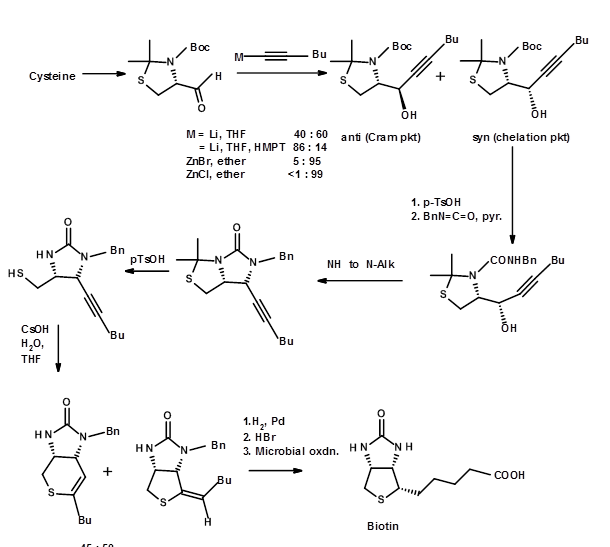
Fig. 4.8.3
In the second example chosen here, the choice of the SM as the appropriate chiron is not obvious but hidden. Such an analysis demands a more critical insight into the concerned stereocentres.
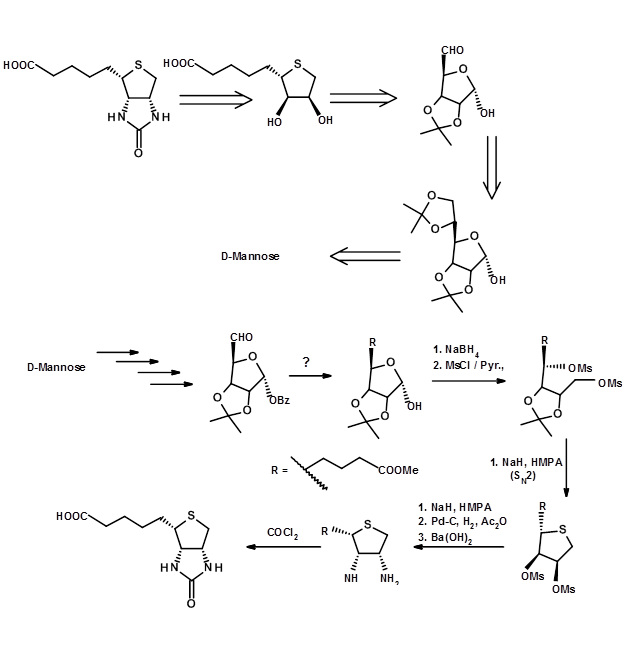
Conclusion
An emerging concept in the Logic In Synthesis is deliberate planning of Green Synthetic Pathways. The logic of retroanalysis is same as discussed above. The only differentiating point is that the criteria for selection of synthetic route discussed earlier would now analyses the same synthetic tree through a Green Chemistry window to select only those routes that have maximum Green aspects. The green chemistry goal is enforced through inclusion the Twelve Principles of Green Chemistry. This could be done by embracing one or more of the following techniques - Use of Green Energy Sources like Microwave, Sonochemistry, Photochemistry etc., solvent free syntheses, using easily recoverable new solvent and eco-friendly solvents, reusable catalysts in syntheses and schemes that avoid protecting group chemistry. Most of the chemistry used in Green Chemistry is not really new to chemists. Chemistry is now revisited due to the environment consciousness that has now crept into industrial chemistry and society at large. Most of the chemistry is buried in two centuries of chemical literature. Several new discoveries in reagents have appeared in recent years. Now Chemists have to become more alert to this awakening to environmental damages caused by chemical activities on this globe.
The above discussions are meant only to illustrate the major steps involved in retrosynthetic analysis of a molecule. Thorough knowledge of synthetic tools, mechanisms and stereochemistry are essential prerequisites for a chemist to venture into the synthesis of complex molecules. Needless to add that all these efforts have to be suitably backed up by a team of chemists, having a rigorous training in laboratory techniques, a first-hand experience on several organic reactions / reagents and thorough knowledge of purification techniques, spectroscopic techniques and not the least, a good knowledge of search techniques to scan and retrieve requisite information from the vast chemical literature accumulated since the dawn of modern chemistry.
Retroanalysis of Some Interesting Molecules
Let us now dwell deep into a few select structures chosen from natural product chemistry and see how these structures have been tackled through different synthetic strategies. We would start with a simple molecule – Disparlure – with only two asymmetric centers. The course would end with a flovour of some Green Chemistry based syntheses to draw the attention of students to this newly emerging concepts and concerns.