
7.1 Nucleophiles and Electrophiles
- Page ID
- 17139
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)Nucleophiles
What is a nucleophile?
Nucleophilic functional groups are those which have electron-rich atoms able to donate a pair of electrons to form a new covalent bond. In both laboratory and biological organic chemistry, the most relevant nucleophilic atoms are oxygen, nitrogen, and sulfur, and the most common nucleophilic functional groups are water, alcohols, phenols, amines, thiols, and occasionally carboxylates.
More specifically in laboratory reactions, halide and azide (N3-) anions are commonly seen acting as nucleophiles.
Of course, carbons can also be nucleophiles - otherwise how could new carbon-carbon bonds be formed in the synthesis of large organic molecules like DNA or fatty acids? Enolate ions (section 7.5) are the most common carbon nucleophiles in biochemical reactions, while the cyanide ion (CN-) is just one example of a carbon nucleophile commonly used in the laboratory. Reactions with carbon nucleophiles will be dealt with in chapters 13 and 14, however - in this chapter and the next, we will concentrate on non-carbon nucleophiles.
When thinking about nucleophiles, the first thing to recognize is that, for the most part, the same quality of 'electron-richness' that makes a something nucleophilic also makes it basic: nucleophiles can be bases, and bases can be nucleophiles. It should not be surprising, then, that most of the trends in basicity that we have already discussed also apply to nucleophilicity.
Now, lets discuss some of the major factors that affect nucleophilicity.
Protonation states and nucleophilicity
The protonation state of a nucleophilic atom has a very large effect on its nucleophilicity. This is an idea that makes intuitive sense: a hydroxide ion is much more nucleophilic (and basic) than a water molecule, because the negatively charged oxygen on the hydroxide ion carries greater electron density than the oxygen atom of a neutral water molecule. In practical terms, this means that a hydroxide nucleophile will react in an SN2 reaction with methyl bromide much faster ( about 10,000 times faster) than a water nucleophile.
Periodic trends and solvent effects in nucleophilicity
There are predictable periodic trends in nucleophilicity. Moving horizontally across the second row of the table, the trend in nucleophilicity parallels the trend in basicity:
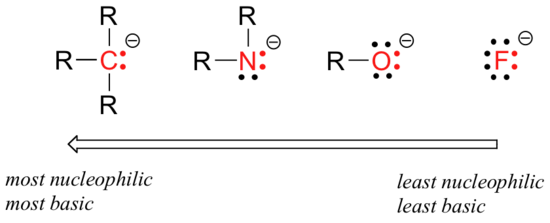
The reasoning behind the horizontal nucleophilicity trend is the same as the reasoning behind the basicity trend: more electronegative elements hold their electrons more tightly, and are less able to donate them to form a new bond.
This horizontal trends also tells us that amines are more nucleophilic than alcohols, although both groups commonly act as nucleophiles in both laboratory and biochemical reactions.
Recall that the basicity of atoms decreases as we move vertically down a column on the periodic table: thiolate ions are less basic than alkoxide ions, for example, and bromide ion is less basic than chloride ion, which in turn is less basic than fluoride ion. Recall also that this trend can be explained by considering the increasing size of the 'electron cloud' around the larger ions: the electron density inherent in the negative charge is spread around a larger area, which tends to increase stability (and thus reduce basicity).
The vertical periodic trend for nucleophilicity is somewhat more complicated that that for basicity: depending on the solvent that the reaction is taking place in, the nucleophilicity trend can go in either direction. Let's take the simple example of the SN2 reaction below:
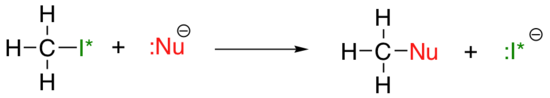
. . .where Nu- is one of the halide ions: fluoride, chloride, bromide, or iodide, and the leaving group I* is a radioactive isotope of iodine (which allows us to distinguish the leaving group from the nucleophile in that case where both are iodide). If this reaction is occurring in a protic solvent (that is, a solvent that has a hydrogen bonded to an oxygen or nitrogen - water, methanol and ethanol are the most important examples), then the reaction will go fastest when iodide is the nucleophile, and slowest when fluoride is the nucleophile, reflecting the relative strength of the nucleophile.

Relative nucleophilicity in a protic solvent
This of course, is opposite that of the vertical periodic trend for basicity, where iodide is the least basic. What is going on here? Shouldn't the stronger base, with its more reactive unbonded valence electrons, also be the stronger nucleophile?
As mentioned above, it all has to do with the solvent. Remember, we are talking now about the reaction running in a protic solvent like ethanol. Protic solvent molecules form very strong ion-dipole interactions with the negatively-charged nucleophile, essentially creating a 'solvent cage' around the nucleophile:

In order for the nucleophile to attack the electrophile, it must break free, at least in part, from its solvent cage. The lone pair electrons on the larger, less basic iodide ion interact less tightly with the protons on the protic solvent molecules - thus the iodide nucleophile is better able to break free from its solvent cage compared the smaller, more basic fluoride ion, whose lone pair electrons are bound more tightly to the protons of the cage.
The picture changes if we switch to a polar aprotic solvent, such as acetone, in which there is a molecular dipole but no hydrogens bound to oxygen or nitrogen. Now, fluoride is the best nucleophile, and iodide the weakest.
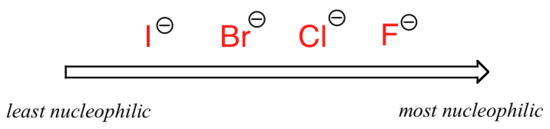
Relative nucleophilicity in a polar aprotic solvent
The reason for the reversal is that, with an aprotic solvent, the ion-dipole interactions between solvent and nucleophile are much weaker: the positive end of the solvent's dipole is hidden in the interior of the molecule, and thus it is shielded from the negative charge of the nucleophile.
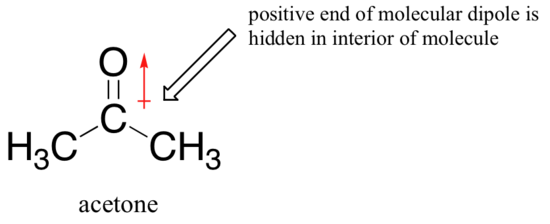
A weaker solvent-nucleophile interaction means a weaker solvent cage for the nucleophile to break through, so the solvent effect is much less important, and the more basic fluoride ion is also the better nucleophile.
Why not use a completely nonpolar solvent, such as hexane, for this reaction, so that the solvent cage is eliminated completely? The answer to this is simple - the nucleophile needs to be in solution in order to react at an appreciable rate with the electrophile, and a solvent such as hexane will not solvate an a charged (or highly polar) nucleophile at all. That is why chemists use polar aprotic solvents for nucleophilic substitution reactions in the laboratory: they are polar enough to solvate the nucleophile, but not so polar as to lock it away in an impenetrable solvent cage. In addition to acetone, three other commonly used polar aprotic solvents are acetonitrile, dimethylformamide (DMF), and dimethyl sulfoxide (DMSO).
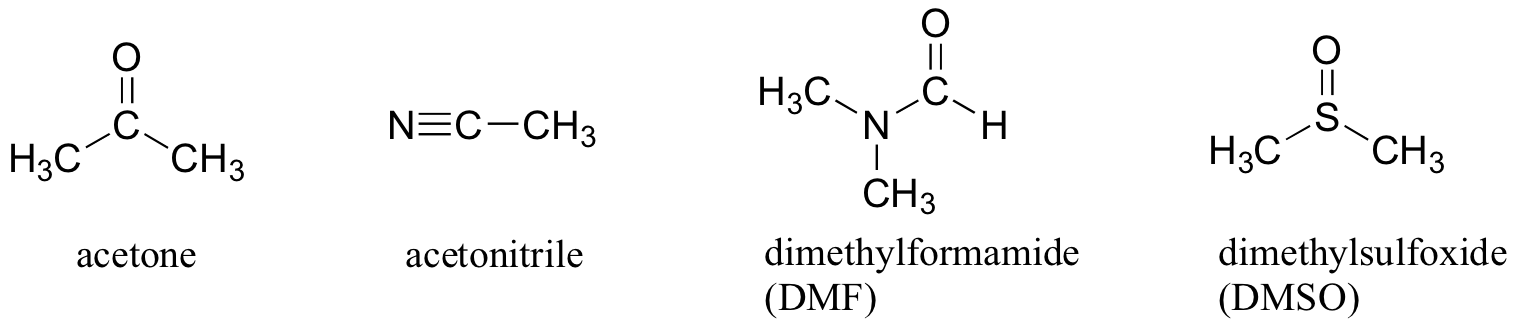
In biological chemistry, where the solvent is protic (water), the most important implication of the periodic trends in nucleophilicity is that thiols are more powerful nucleophiles than alcohols. The thiol group in a cysteine amino acid, for example, is a powerful nucleophile and often acts as a nucleophile in enzymatic reactions, and of course negatively-charged thiolates (RS-) are even more nucleophilic. This is not to say that the hydroxyl groups on serine, threonine, and tyrosine do not also act as nucleophiles - they do.
Resonance effects on nucleophilicity
Resonance effects also come into play when comparing the inherent nucleophilicity of different molecules. The reasoning involved is the same as that which we used to understand resonance effects on basicity. If the electron lone pair on a heteroatom is delocalized by resonance, it is inherently less reactive - meaning less nucleophilic, and also less basic. An alkoxide ion, for example, is more nucleophilic and more basic than a carboxylate group, even though in both cases the nucleophilic atom is a negatively charged oxygen. In the alkoxide, the negative charge is localized on a single oxygen, while in the carboxylate the charge is delocalized over two oxygen atoms by resonance.

The nitrogen atom on an amide is less nucleophilic than the nitrogen of an amine, due to the resonance stabilization of the nitrogen lone pair provided by the amide carbonyl group.

Steric effects on nucleophilicity
Steric hindrance is an important consideration when evaluating nucleophility. For example, tert-butanol is less potent as a nucleophile than methanol. This is because the comparatively bulky methyl groups on the tertiary alcohol effectively block the route of attack by the nucleophilic oxygen, slowing the reaction down considerably (imagine trying to walk through a narrow doorway while carrying three large suitcases!).
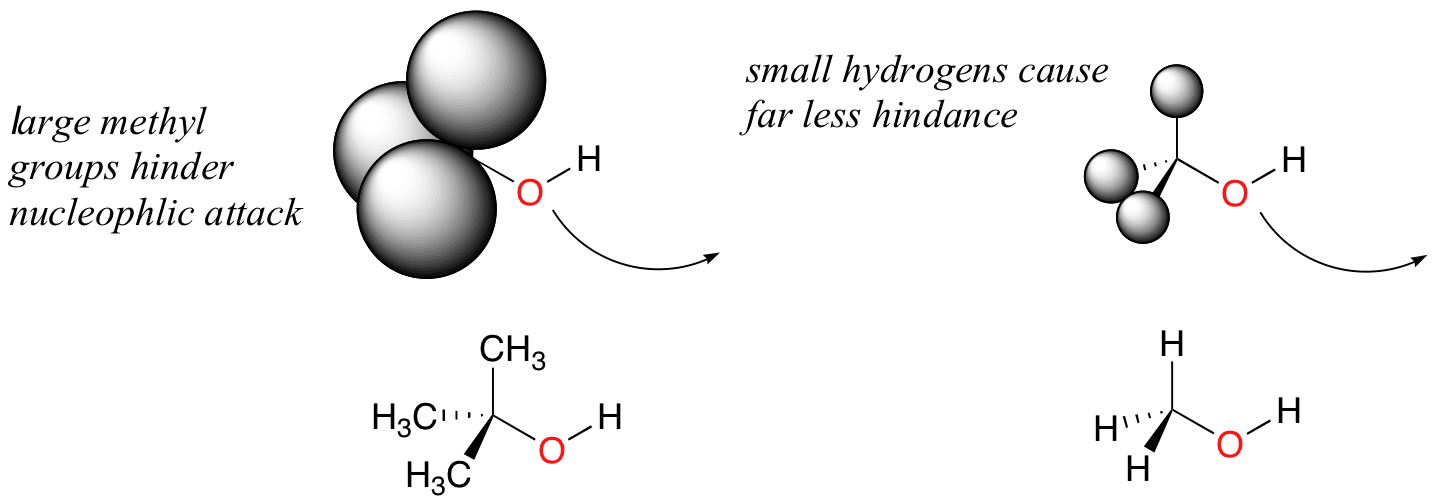
It is not surprising that it is more common to observe serines acting as nucleophiles in enzymatic reactions compared to threonines - the former is a primary alcohol, while the latter is a secondary alcohol.
| Example |
|---|
| Which is the better nucleophile - a cysteine side chain or a methionine side chain? Explain. |
| Example |
|---|
In each of the following pairs of molecules/ions, which is the better nucleophile in a reaction with CH3Br in acetone solvent? Explain your choice.
|
Electrophiles
In the vast majority of the nucleophilic substitution reactions you will see in this and other organic chemistry texts, the electrophilic atom is a carbon which is bonded to an electronegative atom, usually oxygen, nitrogen, sulfur, or a halogen. The concept of electrophilicity is relatively simple: an electron-poor atom is an attractive target for something that is electron-rich, i.e. a nucleophile. However, we must also consider the effect of steric hindrance on electrophilicity. In addition, we must discuss how the nature of the electrophilic carbon, and more specifically the stability of a potential carbocationic intermediate, influences the SN1 vs. SN2 character of a nucleophilic substitution reaction.
Consider two hypothetical SN2 reactions: one in which the electrophile is a methyl carbon and another in which it is tertiary carbon.
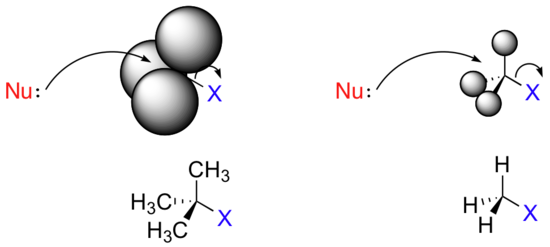
Because the three substituents on the methyl carbon electrophile are tiny hydrogens, the nucleophile has a relatively clear path for backside attack. However, backside attack on the tertiary carbon is blocked by the bulkier methyl groups. Once again, steric hindrance - this time caused by bulky groups attached to the electrophile rather than to the nucleophile - hinders the progress of an associative nucleophilic (SN2) displacement.
The factors discussed in the above paragraph, however, do not prevent a sterically-hindered carbon from being a good electrophile - they only make it less likely to be attacked in a concerted SN2 reaction. Nucleophilic substitution reactions in which the electrophilic carbon is sterically hindered are more likely to occur by a two-step, dissociative (SN1) mechanism. This makes perfect sense from a geometric point of view: the limitations imposed by sterics are significant mainly in an SN2 displacement, when the electrophile being attacked is a sp3-hybridized tetrahedral carbon with its relatively ‘tight’ angles of 109.4o. Remember that in an SN1 mechanism, the nucleophile attacks an sp2-hybridized carbocation intermediate, which has trigonal planar geometry with ‘open’ 120 angles.

With this open geometry, the empty p orbital of the electrophilic carbocation is no longer significantly shielded from the approaching nucleophile by the bulky alkyl groups. A carbocation is a very potent electrophile, and the nucleophilic step occurs very rapidly compared to the first (ionization) step.
Further Reading
MasterOrganicChemistry
Nucleophiles and Electrophiles
What Makes a Good Nucleophile?
The Three Classes of Nucleophile
Carey 4th Edition Online
Khan Academy
Nucleophiles and Electrophiles and the Schwartz Rule
Websites
Electrophiles and Nucleophiles Tutorial
Videos