
11.2: Gaussian Basis Sets
- Page ID
- 210880
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\dsum}{\displaystyle\sum\limits} \)
\( \newcommand{\dint}{\displaystyle\int\limits} \)
\( \newcommand{\dlim}{\displaystyle\lim\limits} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\(\newcommand{\longvect}{\overrightarrow}\)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)A basis set in theoretical and computational chemistry is a set of functions (called basis functions) which are combined in linear combinations (generally as part of a quantum chemical calculation) to create molecular orbitals. For convenience these functions are typically atomic orbitals centered on atoms, but can theoretically be any function; plane waves are frequently used in materials calculations.
The Variational Method and Basis Sets
To describe the electronic states of molecules, we construct wavefunctions for the electronic states by using molecular orbitals. These wavefunctions are approximate solutions to the Schrödinger equation. A mathematical function for a molecular orbital is constructed, \(\psi _i\), as a linear combination of other functions, \(\varphi _j\), which are called basis functions because they provide the basis for representing the molecular orbital.
\[\psi _i = \sum _j c_{ij} \varphi _j \label {10.8}\]
The variational method is used to find values for parameters in the basis functions and for the constant coefficients in the linear combination that optimize these functions, i.e. make them as good as possible. The criterion for quality in the variational method is making the ground state energy of the molecule as low as possible. Here and in the rest of this chapter, the following notation is used: \(\sigma\) is a general spin function (can be either \(\alpha\) or \(\beta\)), \(\varphi \) is the basis function (this usually represents an atomic orbital), \(\psi\) is a molecular orbital, and \(\Psi\) is the electronic state wavefunction (representing a single Slater determinant or linear combination of Slater determinants).
The ultimate goal is a mathematical description of electrons in molecules that enables chemists and other scientists to develop a deep understanding of chemical bonding and reactivity, to calculate properties of molecules, and to make predictions based on these calculations. For example, an active area of research in industry involves calculating changes in chemical properties of pharmaceutical drugs as a result of changes in chemical structure.
Selecting the ab initio model for a chemical system is almost always involves a trade-off between accuracy and computational cost. More accurate methods and larger basis sets make jobs run longer.
In modern computational chemistry, quantum chemical calculations are typically performed using a finite set of basis functions. In these cases, the wavefunctions of the system in question are represented as vectors, the components of which correspond to coefficients in a linear combination of the basis functions in the basis set used.
The molecular spin-orbitals that are used in the Slater determinant usually are expressed as a linear combination of some chosen functions, which are called basis functions. This set of functions is called the basis set. The fact that one function can be represented by a linear combination of other functions is a general property. All that is necessary is that the basis functions span-the-space, which means that the functions must form a complete set and must be describing the same thing. For example, spherical harmonics cannot be used to describe a hydrogen atom radial function because they do not involve the distance r, but they can be used to describe the angular properties of anything in three-dimensional space.
This span-the-space property of functions is just like the corresponding property of vectors. The unit vectors \((\overrightarrow {x}, \overrightarrow {y}, \overrightarrow {z})\) describe points in space and form a complete set since any position in space can be specified by a linear combination of these three unit vectors. These unit vectors also could be called basis vectors.
Explain why the unit vectors \((\overrightarrow {x}, \overrightarrow {y})\) do not form a complete set to describe your (three-dimensional) classroom.
Just as we discussed for atoms, parameters in the basis functions and the coefficients in the linear combination can be optimized in accord with the Variational Principle to produce a self-consistent field (SCF) for the electrons. This optimization means that the ground state energy calculated with the wavefunction is minimized with respect to variation of the parameters and coefficients defining the function. As a result, that ground state energy is larger than the exact energy, but is the best value that can be obtained with that wavefunction.
Slater Type Orbitals (STOs)
Intuitively one might select hydrogenic atomic orbitals as the basis set for molecular orbitals. After all, molecules are composed of atoms, and hydrogenic orbitals describe atoms exactly if the electron-electron interactions are neglected. At a better level of approximation, the nuclear charge that appears in these functions can be used as a variational parameter to account for the shielding effects due to the electron-electron interactions. Also, the use of atomic orbitals allows us to interpret molecular properties and charge distributions in terms of atomic properties and charges, which is very appealing since we picture molecules as composed of atoms. As described in the previous chapter, calculations with hydrogenic functions were not very efficient so other basis functions, Slater-type atomic orbitals (STOs), were invented.
A minimal basis set of STOs for a molecule includes only those STOs that would be occupied by electrons in the atoms forming the molecule. A larger basis set, however, improves the accuracy of the calculations by providing more variable parameters to produce a better approximate wavefunction, but at the expense of increased computational time. STOs have the following radial part (the spherical harmonic functions are used to describe the angular part)
\[R(r) = N r^{n − 1} e^{−\zeta r}\]
where
- \(n\) is a natural number that plays the role of principal quantum number, n = 1,2,...,
- \(N\) is a normalizing constant,
- \(r\) is the distance of the electron from the atomic nucleus, and \(\zeta\) is a constant related to the effective charge of the nucleus, the nuclear charge being partly shielded by electrons. Historically, the effective nuclear charge was estimated by Slater's rules.
Double-zeta basis Sets
One can use more than one STO to represent one atomic orbital, as shown in Equation \(\ref{10.11}\), and rather than doing a nonlinear variational calculation to optimize each \(\zeta\) value, use two STOs with different \(\zeta\) variables. The linear variation calculation then will produce the coefficients (\(C_1\) and \(C_2\)) for these two functions in the linear combination that best describes the charge distribution in the molecule (for the ground state). The function with the large zeta accounts for charge near the nucleus, while the function with the smaller zeta accounts for the charge distribution at larger values of the distance from the nucleus. This expanded basis set is called a double-zeta basis set.
\[R_{2s} (r) = C_1re^{-\zeta _1r} + C_2 r e^{-\zeta _2 r} \label {10.11}\]
The use of double zeta functions in basis sets is especially important because without them orbitals of the same type are constrained to be identical even though in the molecule they may be chemically inequivalent. For example, in acetylene the \(p_z\) orbital along the internuclear axis is in a quite different chemical environment and is being used to account for quite different bonding than the \(p_x\) and \(p_y\) orbitals. With a double zeta basis set the \(p_z\) orbital is not constrained to be the same size as the \(p_x\) and \(p_y\) orbitals.
Explain why the \(p_x\), \(p_y\), and \(p_z\) orbitals in a molecule might be constrained to be the same in a single-zeta basis set calculation, and how the use of a double-zeta basis set would allow the \(p_x\), \(p_y\), and \(p_z\) orbitals to differ.
Gaussian Orbitals
Although any basis set that sufficiently spans the space of electron distribution could be used, the concept of Molecular Orbitals as Linear Combinations of Atomic Orbitals (LCAO) suggests a very natural set of basis functions: AO-type functions centered on each nuclei. One obvious choice are the exact hydrogen AO's, known as Slater-type orbitals (STO)--describing the radial component of the functions. However, the computation of the integrals is greatly simplified by using Gaussian-type orbitals (GTO) for basis functions.
While the STO basis set was an improvement over hydrogenic orbitals in terms of computational efficiency, representing the STOs with Gaussian functions produced further improvements that were needed to accurately describe molecules. A Gaussian basis function has the form shown in Equation \(\ref{10.12}\). Note that in all the basis sets, only the radial part of the orbital changes, and the spherical harmonic functions are used in all of them to describe the angular part of the orbital.
\[ G_{nlm} (r, \theta , \psi ) = N_n \underbrace{r^{n-1} e^{-\alpha r^2}}_{\text{radial part}} \underbrace{Y^m_l (\theta, \psi)}_{\text{angular part}} \label{10.12}\]
Unfortunately Gaussian functions do not match the shape of an atomic orbital very well. In particular, they are flat rather than steep near the atomic nucleus at \(r = 0\), and they fall off more rapidly at large values of \(r\) (Figure \(\PageIndex{1}\)).
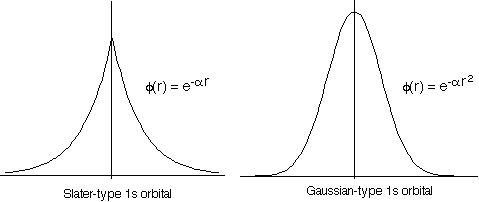
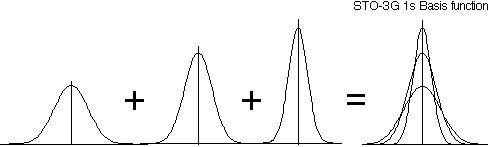
To compensate for this problem, each STO is replaced with a number of Gaussian functions with different values for the exponential parameter. These Gaussian functions form a primitive Gaussian basis set. Linear combinations of the primitive Gaussians are formed to approximate the radial part of an STO. This linear combination is not optimized further in the energy variational calculation, but rather is frozen and treated as a single function. The linear combination of primitive Gaussian functions is called a contracted Gaussian function. Although more functions and more integrals now are part of the calculation, the integrals involving Gaussian functions are quicker to compute than those involving exponentials, so there is a net gain in the efficiency of the calculation.
Gaussian basis sets are identified by abbreviations such as N-MPG*. N is the number of Gaussian primitives used for each inner-shell orbital. The hyphen indicates a split-basis set where the valence orbitals are double zeta. The M indicates the number of primitives that form the large zeta function (for the inner valence region), and P indicates the number that form the small zeta function (for the outer valence region). G identifies the set a being Gaussian. The addition of an asterisk to this notation means that a single set of Gaussian 3d polarization functions (discussed elswhere) is included. A double asterisk means that a single set of Gaussian 2p functions is included for each hydrogen atom.
For example, 3G means each STO is represented by a linear combination of three primitive Gaussian functions. 6-31G means each inner shell (1s orbital) STO is a linear combination of 6 primitives and each valence shell STO is split into an inner and outer part (double zeta) using 3 and 1 primitive Gaussians, respectively (see Table \(\PageIndex{1}\) for other examples).
| Basis set | # functions | Basis set | # functions | Basis set | # functions | ||
|---|---|---|---|---|---|---|---|
| STO-3G | 5 | 6-31G | 9 | 6-311G | 13 | ||
| 3-21G | 9 | 6-31G* | 15 | 6-311G* | 18* | ||
| 4-31G | 9 | 6-31+G* | 19 | 6-311+G* | 22* |
The 1s Slater-type orbital \(S_1 (r) = \sqrt {4 \zeta _1 e^{-\zeta _1 r}}\) with \(\zeta _1 = 1.24 \) is represented as a sum of three primitive Gaussian functions,
\[S_G (r) = \sum _{j=1}^3 C_j e^{-\alpha _j r^2} \nonumber \]
This sum is the contracted Gaussian function for the STO.
- Make plots of the STO and the contracted Gaussian function on the same graph so they can be compared easily. All distances should be in units of the Bohr radius. Use the following values for the coefficients, C, and the exponential parameters, \(\alpha\).
index j \(\alpha _j\) \(C_j\) 1 0.1688 0.4 2 0.6239 0.7 3 3.425 1.3 - Change the values of the coefficients and exponential parameters to see if a better fit can be obtained.
- Comment on the ability of a linear combination of Gaussian functions to accurately describe a STO.
Summary
When molecular calculations are performed, it is common to use a basis composed of a finite number of atomic orbitals (Equation \(\ref{10.8}\)), centered at each atomic nucleus within the molecule (linear combination of atomic orbitals ansatz). These atomic orbitals are well described with Slater-type orbitals (STOs), as STOs decay exponentially with distance from the nuclei, accurately describing the long-range overlap between atoms, and reach a maximum at zero, well describing the charge and spin at the nucleus. STOs are computationally difficult and it was later realized by Frank Boys that these Slater-type orbitals could in turn be approximated as linear combinations of Gaussian orbitals instead. Because it is easier to calculate overlap and other integrals with Gaussian basis functions, this led to huge computational savings
Contributors
- Wikipedia
David M. Hanson, Erica Harvey, Robert Sweeney, Theresa Julia Zielinski ("Quantum States of Atoms and Molecules")