
9.E: Titrimetric Methods (Exercises)
- Page ID
- 70695
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)Some of the problems that follow require one or more equilibrium constants or standard state potentials. For your convenience, here are hyperlinks to the appendices containing these constants.
Appendix 10: Solubility Products
Appendix 11: Acid Dissociation Constants
Appendix 12: Metal-Ligand Formation Constants
Appendix 13: Standard State Reduction Potentials
1. Calculate or sketch titration curves for the following acid–base titrations.
- 25.0 mL of 0.100 M NaOH with 0.0500 M HCl
- 50.0 mL of 0.0500 M HCOOH with 0.100 M NaOH
- 50.0 mL of 0.100 M NH3 with 0.100 M HCl
- 50.0 mL of 0.0500 M ethylenediamine with 0.100 M HCl
- 50.0 mL of 0.0400 M citric acid with 0.120 M NaOH
- 50.0 mL of 0.0400 M H3PO4 with 0.120 M NaOH
2. Locate the equivalence point for each titration curve in problem 1. What is the stoichiometric relationship between the moles of acid and the moles of base at each of these equivalence points?
3. Suggest an appropriate visual indicator for each of the titrations in problem 1.
4. In sketching the titration curve for a weak acid we approximate the pH at 10% of the equivalence point volume as pKa – 1, and the pH at 90% of the equivalence point volume as pKa + 1. Show that these assumptions are reasonable.
5. Tartaric acid, H2C4H4O6, is a diprotic weak acid with a pKa1 of 3.0 and a pKa2 of 4.4. Suppose you have a sample of impure tartaric acid (purity > 80%), and that you plan to determine its purity by titrating with a solution of 0.1 M NaOH using an indicator to signal the end point. Describe how you will carry out the analysis, paying particular attention to how much sample to use, the desired pH range for the indicator, and how you will calculate the %w/w tartaric acid.
6. The following data for the titration of a monoprotic weak acid with a strong base were collected using an automatic titrator. Prepare normal, first derivative, second derivative, and Gran plot titration curves for this data, and locate the equivalence point for each.
|
Volume of NaOH (ml) |
pH |
Volume of NaOH (mL) |
pH |
|
0.25 |
3.0 |
49.95 |
7.8 |
|
0.86 |
3.2 |
49.97 |
8.0 |
|
1.63 |
3.4 |
49.98 |
8.2 |
|
2.72 |
3.6 |
49.99 |
8.4 |
|
4.29 |
3.8 |
50.00 |
8.7 |
|
6.54 |
4.0 |
50.01 |
9.1 |
|
9.67 |
4.2 |
50.02 |
9.4 |
|
13.79 |
4.4 |
50.04 |
9.6 |
|
18.83 |
4.6 |
50.06 |
9.8 |
|
24.47 |
4.8 |
50.10 |
10.0 |
|
30.15 |
5.0 |
50.16 |
10.2 |
|
35.33 |
5.2 |
50.25 |
10.4 |
|
39.62 |
5.4 |
50.40 |
10.6 |
|
42.91 |
5.6 |
50.63 |
10.8 |
|
45.28 |
5.8 |
51.01 |
11.0 |
|
46.91 |
6.0 |
51.61 |
11.2 |
|
48.01 |
6.2 |
52.58 |
11.4 |
|
48.72 |
6.4 |
54.15 |
11.6 |
|
49.19 |
6.6 |
56.73 |
11.8 |
|
49.48 |
6.8 |
61.11 |
12.0 |
|
49.67 |
7.0 |
68.83 |
12.2 |
|
49.79 |
7.2 |
83.54 |
12.4 |
|
49.78 |
7.4 |
116.14 |
12.6 |
|
49.92 |
7.6 |
7. Schwartz published the following simulated data for the titration of a 1.02 × 10–4 M solution of a monoprotic weak acid (pKa = 8.16) with 1.004 × 10–3 M NaOH.10 The simulation assumes that a 50-mL pipet is used to transfer a portion of the weak acid solution to the titration vessel. A calibration of the pipet shows that it delivers a volume of only 49.94 mL. Prepare normal, first derivative, second derivative, and Gran plot titration curves for this data, and determine the equivalence point for each. How do these equivalence points compare to the expected equivalence point? Comment on the utility of each titration curve for the analysis of very dilute solutions of very weak acids.
|
mL of NaOH |
pH |
mL of NaOH |
pH |
|
0.03 |
6.212 |
4.79 |
8.858 |
|
0.09 |
6.504 |
4.99 |
8.926 |
|
0.29 |
6.936 |
5.21 |
8.994 |
|
0.72 |
7.367 |
5.41 |
9.056 |
|
1.06 |
7.567 |
5.61 |
9.118 |
|
1.32 |
7.685 |
5.85 |
9.180 |
|
1.53 |
7.776 |
6.05 |
9.231 |
|
1.76 |
7.863 |
6.28 |
9.283 |
|
1.97 |
7.938 |
6.47 |
9.327 |
|
2.18 |
8.009 |
6.71 |
9.374 |
|
2.38 |
8.077 |
6.92 |
9.414 |
|
2.60 |
8.146 |
7.15 |
9.451 |
|
2.79 |
8.208 |
7.36 |
9.484 |
|
3.01 |
8.273 |
7.56 |
9.514 |
|
3.41 |
8.332 |
7.79 |
9.545 |
|
3.60 |
8.458 |
8.21 |
9.572 |
|
3.80 |
8.521 |
8.44 |
9.599 |
|
3.99 |
8.584 |
8.64 |
9.645 |
|
4.18 |
8.650 |
8.84 |
9.666 |
|
4.40 |
8.720 |
9.07 |
9.688 |
|
4.57 |
8.784 |
9.27 |
9.706 |
8. Calculate or sketch the titration curve for a 50.0 mL solution of a 0.100 M monoprotic weak acid (pKa = 8) with 0.1 M strong base in a nonaqueous solvent with Ks = 10–20. You may assume that the change in solvent does not affect the weak acid’s pKa. Compare your titration curve to the titration curve when water is the solvent.
9. The titration of a mixture of p-nitrophenol (pKa = 7.0) and m-nitrophenol (pKa = 8.3) can be followed spectrophotometrically. Neither acid absorbs at a wavelength of 545 nm, but their respective conjugate bases do absorb at this wavelength. The m-nitrophenolate ion has a greater absorbance than an equimolar solution of the p-nitrophenolate ion. Sketch the spectrophotometric titration curve for a 50.00-mL mixture consisting of 0.0500 M p-nitrophenol and 0.0500 M m-nitrophenol with 0.100 M NaOH. Compare your result to the expected potentiometric titration curves.
10. The quantitative analysis for aniline (C6H5NH2, Kb = 3.94 × 10–10) can be carried out by an acid–base titration using glacial acetic acid as the solvent and HClO4 as the titrant. A known volume of sample containing 3–4 mmol of aniline is transferred to a 250-mL Erlenmeyer flask and diluted to approximately 75 mL with glacial acetic acid. Two drops of a methyl violet indicator are added, and the solution is titrated with previously standardized 0.1000 M HClO4 (prepared in glacial acetic acid using anhydrous HClO4) until the end point is reached. Results are reported as parts per million aniline.
(a) Explain why this titration is conducted using glacial acetic acid as the solvent instead of water.
(b) One problem with using glacial acetic acid as solvent is its relatively high coefficient of thermal expansion of 0.11%/oC. For example, 100.00 mL of glacial acetic acid at 25oC occupies 100.22 mL at 27oC. What is the effect on the reported concentration of aniline if the standardization of HClO4 is conducted at a temperature that is lower than that for the analysis of the unknown?
(c) The procedure calls for a sample containing 3–4 mmoles of aniline. Why is this requirement necessary?
11. Using a ladder diagram, explain why the presence of dissolved CO2 leads to a determinate error for the standardization of NaOH if the end point’s pH falls between 6–10, but no determinate error if the end point’s pH is less than 6.
12. A water sample’s acidity is determined by titrating to fixed end point pHs of 3.7 and 8.3, with the former providing a measure of the concentration of strong acid, and the later a measure of the combined concentrations of strong acid and weak acid. Sketch a titration curve for a mixture of 0.10 M HCl and 0.10 M H2CO3 with 0.20 M strong base, and use it to justify the choice of these end points.
13. Ethylenediaminetetraacetic acid, H4Y, is a weak acid with successive acid dissociation constants of 0.010, 2.19 × 10–3, 6.92 × 10–7, and 5.75 × 10–11. Figure 9.46 shows a titration curve for H4Y with NaOH. What is the stoichiometric relationship between H4Y and NaOH at the equivalence point marked with the red arrow?
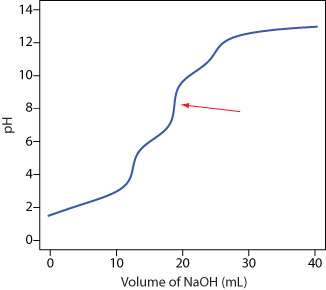
Figure 9.46 Titration curve for Problem 9.13.
14. A Gran plot method has been described for the quantitative analysis of a mixture consisting of a strong acid and a monoprotic weak acid.11 A 50.00-mL mixture of HCl and CH3COOH is transferred to an Erlenmeyer flask and titrated by using a digital pipet to add successive 1.00-mL aliquots of 0.09186 M NaOH. The progress of the titration is monitored by recording the pH after each addition of titrant. Using the two papers listed in the footnote as a reference, prepare a Gran plot for the following data, and determine the concentrations of HCl and CH3COOH.
|
Volume of |
|
Volume of |
|
Volume of |
|
|
1.00 |
1.83 |
24.00 |
4.45 |
47.00 |
12.14 |
|
2.00 |
1.86 |
25.00 |
4.53 |
48.00 |
12.17 |
|
3.00 |
1.89 |
26.00 |
4.61 |
49.00 |
12.20 |
|
4.00 |
1.92 |
27.00 |
4.69 |
50.00 |
12.23 |
|
5.00 |
1.95 |
28.00 |
4.76 |
51.00 |
12.26 |
|
6.00 |
1.99 |
29.00 |
4.84 |
52.00 |
12.28 |
|
7.00 |
2.03 |
30.00 |
4.93 |
53.00 |
12.30 |
|
8.00 |
2.10 |
31.00 |
5.02 |
54.00 |
12.32 |
|
9.00 |
2.18 |
32.00 |
5.13 |
55.00 |
12.34 |
|
10.00 |
2.31 |
33.00 |
5.23 |
56.00 |
12.36 |
|
11.00 |
2.51 |
34.00 |
5.37 |
57.00 |
12.38 |
|
12.00 |
2.81 |
35.00 |
5.52 |
58.00 |
12.39 |
|
13.00 |
3.16 |
36.00 |
5.75 |
59.00 |
12.40 |
|
14.00 |
3.36 |
37.00 |
6.14 |
60.00 |
12.42 |
|
15.00 |
3.54 |
38.00 |
10.30 |
61.00 |
12.43 |
|
16.00 |
3.69 |
39.00 |
11.31 |
62.00 |
12.44 |
|
17.00 |
3.81 |
40.00 |
11.58 |
63.00 |
12.45 |
|
18.00 |
3.93 |
41.00 |
11.74 |
64.00 |
12.47 |
|
19.00 |
4.02 |
42.00 |
11.85 |
65.00 |
12.48 |
|
20.00 |
4.14 |
43.00 |
11.93 |
66.00 |
12.49 |
|
21.00 |
4.22 |
44.00 |
12.00 |
67.00 |
12.50 |
|
22.00 |
4.30 |
45.00 |
12.05 |
68.00 |
12.51 |
|
23.00 |
4.38 |
46.00 |
12.10 |
69.00 |
12.52 |
15. Explain why it is not possible for a sample of water to simultaneously have OH– and HCO3– as sources of alkalinity.
16. For each of the following, determine the sources of alkalinity (OH–, HCO3–, CO32–) and their respective concentrations in parts per million. In each case a 25.00-mL sample is titrated with 0.1198 M HCl to the bromocresol green and the phenolphthalein end points.
|
Volume of HCl (mL) to the |
Volume of HCl (mL) to the |
|||
| a |
21.36 |
21.38 |
||
| b |
5.67 |
21.13 |
||
| c |
0.00 |
14.28 |
||
| d |
17.12 |
34.26 |
||
| e |
21.36 |
25.69 |
||
17. A sample may contain any of the following: HCl, NaOH, H3PO4, H2PO4–, HPO42–, or PO43–. The composition of a sample is determined by titrating a 25.00-mL portion with 0.1198 M HCl or 0.1198 M NaOH to the phenolphthalein and the methyl orange end points. For each of the following, determine which species are present in the sample, and their respective molar concentrations.
| Titrant |
Phenolphthalein end |
methyl orange end point |
||||
| a | HCl |
11.54 |
35.29 |
|||
| b | NaOH |
19.79 |
9.89 |
|||
| c | HCl |
22.76 |
22.78 |
|||
| d | NaOH |
39.42 |
17.48 |
|||
18. The protein in a 1.2846-g sample of an oat cereal is determined by a Kjeldahl analysis. The sample is digested with H2SO4, the resulting solution made basic with NaOH, and the NH3 distilled into 50.00 mL of 0.09552 M HCl. The excess HCl is back titrated using 37.84 mL of 0.05992 M NaOH. Given that the proteins in grains average 17.54% w/w N, report the %w/w protein in the sample.
19. The concentration of SO2 in air is determined by bubbling a sample of air through a trap containing H2O2. Oxidation of SO2 by H2O2 results in the formation of H2SO4, which is then determined by titrating with NaOH. In a typical analysis, a sample of air was passed through the peroxide trap at a rate of 12.5 L/min for 60 min and required 10.08 mL of 0.0244 M NaOH to reach the phenolphthalein end point. Calculate the μL/L SO2 in the sample of air. The density of SO2 at the temperature of the air sample is 2.86 mg/mL.
20. The concentration of CO2 in air is determined by an indirect acid–base titration. A sample of air is bubbled through a solution containing an excess of Ba(OH)2, precipitating BaCO3. The excess Ba(OH)2 is back titrated with HCl. In a typical analysis a 3.5-L sample of air was bubbled through 50.00 mL of 0.0200 M Ba(OH)2. Back titrating with 0.0316 M HCl required 38.58 mL to reach the end point. Determine the ppm CO2 in the sample of air given that the density of CO2 at the temperature of the sample is 1.98 g/L.
21. The purity of a synthetic preparation of methylethyl ketone, C3H8O, is determined by reacting it with hydroxylamine hydrochloride, liberating HCl (see reaction in Table 9.8). In a typical analysis a 3.00-mL sample was diluted to 50.00 mL and treated with an excess of hydroxylamine hydrochloride. The liberated HCl was titrated with 0.9989 M NaOH, requiring 32.68 mL to reach the end point. Report the percent purity of the sample given that the density of methylethyl ketone is 0.805 g/mL.
22. Animal fats and vegetable oils are triesters formed from the reaction between glycerol (1,2,3-propanetriol) and three long-chain fatty acids. One of the methods used to characterize a fat or an oil is a determination of its saponification number. When treated with boiling aqueous KOH, an ester saponifies into the parent alcohol and fatty acids (as carboxylate ions). The saponification number is the number of milligrams of KOH required to saponify 1.000 gram of the fat or the oil. In a typical analysis a 2.085-g sample of butter is added to 25.00 mL of 0.5131 M KOH. After saponification is complete the excess KOH is back titrated with 10.26 mL of 0.5000 M HCl. What is the saponification number for this sample of butter?
23. A 250.0-mg sample of an organic weak acid is dissolved in an appropriate solvent and titrated with 0.0556 M NaOH, requiring 32.58 mL to reach the end point. Determine the compound’s equivalent weight.
24. Figure 9.47 shows a potentiometric titration curve for a 0.4300-g sample of a purified amino acid that was dissolved in 50.00 mL of water and titrated with 0.1036 M NaOH. Identify the amino acid from the possibilities listed in the following table.
| amino acid | formula weight (g/mol) | Ka |
| alanine | 89.1 | 1.36 × 10–10 |
| glycine | 75.1 | 1.67 × 10–10 |
| methionine | 149.2 | 8.9 × 10–10 |
| taurine | 125.2 | 1.8 × 10–9 |
| asparagine | 150 | 1.9 × 10–9 |
| leucine | 131.2 | 1.79 × 10–10 |
| phenylalanine | 166.2 | 4.9 × 10–10 |
| valine | 117.2 | 1.91 × 10–10 |
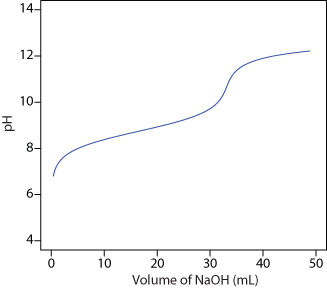
Figure 9.47 Titration curve for Problem 9.24.
25. Using its titration curve, determine the acid dissociation constant for the weak acid in problem 9.6.
26. Where in the scale of operations do the microtitration techniques discussed in section 9B.7 belong?
27. An acid–base titration may be used to determine an analyte’s gram equivalent weight, but it can not be used to determine its gram formula weight. Explain why.
28. Commercial washing soda is approximately 30–40% w/w Na2CO3. One procedure for the quantitative analysis of washing soda contains the following instructions:
Transfer an approximately 4-g sample of the washing soda to a 250-mL volumetric flask. Dissolve the sample in about 100 mL of H2O and then dilute to the mark. Using a pipet, transfer a 25-mL aliquot of this solution to a 125-mL Erlenmeyer flask, and add 25-mL of H2O and 2 drops of bromocresol green indicator. Titrate the sample with 0.1 M HCl to the indicator’s end point.
What modifications, if any, are necessary if you want to adapt this procedure to evaluate the purity of commercial Na2CO3 that is >98% pure?
29. A variety of systematic and random errors are possible when standardizing a solution of NaOH against the primary weak acid standard potassium hydrogen phthalate (KHP). Identify, with justification, whether the following are systematic or random sources of error, or if they have no effect. If the error is systematic, then indicate whether the experimentally determined molarity for NaOH is too high or too low. The standardization reaction is
\[\ce{C_8H_5O_4^-}(aq)+\mathrm{OH^-}(aq)\rightarrow \mathrm{C_8H_4O_4^{2-}}(aq)+\mathrm{H_2O}(l)\]
(a) The balance used to weigh KHP is not properly calibrated and always reads 0.15 g too low.
(b) The indicator for the titration changes color between a pH of 3–4.
(c) An air bubble, which is lodged in the buret’s tip at the beginning of the analysis, dislodges during the titration.
(d) Samples of KHP are weighed into separate Erlenmeyer flasks, but the balance is only tarred with the first flask.
(e) The KHP is not dried before it was used.
(f) The NaOH is not dried before it was used.
(g) The procedure states that the sample of KHP should be dissolved in 25 mL of water, but it is accidentally dissolved in 35 mL of water.
30. The concentration of o-phthalic acid in an organic solvent, such as n-butanol, is determined by an acid–base titration using aqueous NaOH as the titrant. As the titrant is added, the o-phthalic acid is extracted into the aqueous solution where it reacts with the titrant. The titrant must be added slowly to allow sufficient time for the extraction to take place.
(a) What type of error do you expect if the titration is carried out too quickly?
(b) Propose an alternative acid–base titrimetric method that allows for a more rapid determination of the concentration of o-phthalic acid in n‑butanol.
31. Calculate or sketch titration curves for 50.00 mL of 0.0500 Mg2+ with 0.0500 M EDTA at a pH of 7 and 10. Locate the equivalence point for each titration curve.
32. Calculate or sketch titration curves for 25.0 mL of 0.0500 M Cu2+ with 0.025 M EDTA at a pH of 10, and in the presence of 10–3 M and 10–1 M NH3. Locate the equivalence point for each titration curve.
33. Sketch the spectrophotometric titration curve for the titration of a mixture of 5.00 × 10–3 M Bi3+ and 5.00 × 10–3 M Cu2+ with 0.0100 M EDTA. Assume that only the Cu2+–EDTA complex absorbs at the selected wavelength.
34. The EDTA titration of mixtures of Ca2+ and Mg2+ can be followed thermometrically because the formation of the Ca2+–EDTA complex is exothermic and the formation of the Mg2+–EDTA complex is endothermic. Sketch the thermometric titration curve for a mixture of 5.00 × 10–3 M Ca2+ and 5.00 × 10–3 M Mg2+ with 0.0100 M EDTA. The heats of formation for CaY2– and MgY2– are, respectively, –23.9 kJ/mole and 23.0 kJ/mole.
35. EDTA is one member of a class of aminocarboxylate ligands that form very stable 1:1 complexes with metal ions. The following table provides logKf values for the complexes of six such ligands with Ca2+ and Mg2+. Which ligand is the best choice for the direct titration of Ca2+ in the presence of Mg2+?
|
Mg2+ |
Ca2+ |
||
|
EDTA |
ethylenediaminetetraacetic acid |
8.7 |
10.7 |
|
HEDTA |
N-hydroxyethylenediaminetriacetic acid |
7.0 |
8.0 |
|
EEDTA |
ethyletherdiaminetetraacetic acid |
8.3 |
10.0 |
|
DGTA |
ethyleneglycol-bis(β-aminoethylether)- |
5.4 |
10.9 |
|
DTPA |
diethylenetriaminepentaacetic acid |
9.0 |
10.7 |
|
CyDTA |
cyclohexanediaminetetraacetic acid |
10.3 |
12.3 |
36. The amount of calcium in physiological fluids can be determined by a complexometric titration with EDTA. In one such analysis a 0.100-mL sample of a blood serum was made basic by adding 2 drops of NaOH and titrated with 0.00119 M EDTA, requiring 0.268 mL to reach the end point. Report the concentration of calcium in the sample as milligrams Ca per 100 mL.
37. After removing the membranes from an eggshell, the shell is dried and its mass recorded as 5.613 g. The eggshell is transferred to a 250-mL beaker and dissolved in 25 mL of 6 M HCl. After filtering, the solution containing the dissolved eggshell is diluted to 250 mL in a volumetric flask. A 10.00-mL aliquot is placed in a 125-mL Erlenmeyer flask and buffered to a pH of 10. Titrating with 0.04988 M EDTA requires 44.11 mL to reach the end point. Determine the amount of calcium in the eggshell as %w/w CaCO3.
38. The concentration of cyanide, CN–, in a copper electroplating bath can be determined by a complexometric titration with Ag+, forming the soluble Ag(CN)2– complex. In a typical analysis a 5.00-mL sample from an electroplating bath is transferred to a 250-mL Erlenmeyer flask, and treated with 100 mL of H2O, 5 mL of 20% w/v NaOH and 5 mL of 10% w/v KI. The sample is titrated with 0.1012 M AgNO3, requiring 27.36 mL to reach the end point as signaled by the formation of a yellow precipitate of AgI. Report the concentration of cyanide as parts per million of NaCN.
39. Before the introduction of EDTA most complexation titrations used Ag+ or CN– as the titrant. The analysis for Cd2+, for example, was accomplished indirectly by adding an excess of KCN to form Cd(CN)42–, and back titrating the excess CN– with Ag+, forming Ag(CN)2–. In one such analysis a 0.3000-g sample of an ore was dissolved and treated with 20.00 mL of 0.5000 M KCN. The excess CN– required 13.98 mL of 0.1518 M AgNO3 to reach the end point. Determine the %w/w Cd in the ore.
40. Solutions containing both Fe3+ and Al3+ can be selectively analyzed for Fe3+ by buffering to a pH of 2 and titrating with EDTA. The pH of the solution is then raised to 5 and an excess of EDTA added, resulting in the formation of the Al3+–EDTA complex. The excess EDTA is back-titrated using a standard solution of Fe3+, providing an indirect analysis for Al3+.
(a) At a pH of 2, verify that the formation of the Fe3+–EDTA complex is favorable, and that the formation of the Al3+–EDTA complex is not favorable.
(b) A 50.00-mL aliquot of a sample containing Fe3+ and Al3+ is transferred to a 250-mL Erlenmeyer flask and buffered to a pH of 2. A small amount of salicylic acid is added, forming the soluble red-colored Fe3+–salicylic acid complex. The solution is titrated with 0.05002 M EDTA, requiring 24.82 mL to reach the end point as signaled by the disappearance of the Fe3+–salicylic acid complex’s red color. The solution is buffered to a pH of 5 and 50.00 mL of 0.05002 M EDTA is added. After ensuring that the formation of the Al3+–EDTA complex is complete, the excess EDTA was back titrated with 0.04109 M Fe3+, requiring 17.84 mL to reach the end point as signaled by the reappearance of the red-colored Fe3+–salicylic acid complex. Report the molar concentrations of Fe3+ and Al3+ in the sample.
41. Prada and colleagues described an indirect method for determining sulfate in natural samples, such as seawater and industrial effluents.12 The method consists of three steps: precipitating the sulfate as PbSO4; dissolving the PbSO4 in an ammonical solution of excess EDTA to form the soluble PbY2– complex; and titrating the excess EDTA with a standard solution of Mg2+. The following reactions and equilibrium constants are known
|
\(\mathrm{PbSO_4}(s)\rightleftharpoons \mathrm{Pb^{2+}}(aq)+\mathrm{SO_4^{2-}}(aq)\) |
\(K_\textrm{sp}=1.6\times10^{-8}\) |
|
\(\mathrm{Pb^{2+}}(aq)+\mathrm{Y^{4-}}(aq)\rightleftharpoons\mathrm{PbY^{2-}}(aq)\) |
\(K_\textrm f=1.1\times10^{18}\) |
|
\(\mathrm{Mg^{2+}}(aq)+\mathrm{Y^{4-}}(aq)\rightleftharpoons\mathrm{MgY^{2-}}(aq)\) |
\(K_\textrm f=4.9\times10^8\) |
|
\(\mathrm{Zn^{2+}}(aq)+\mathrm{Y^{4-}}(aq)\rightleftharpoons \mathrm{ZnY^{2-}}(aq)\) |
\(K_\textrm f=3.2\times10^{16}\) |
(a) Verify that a precipitate of PbSO4 dissolves in a solution of Y4–.
(b) Sporek proposed a similar method using Zn2+ as a titrant and found that the accuracy was frequently poor.13 One explanation is that Zn2+ might react with the PbY2–complex, forming ZnY2–. Show that this might be a problem when using Zn2+ as a titrant, but that it is not a problem when using Mg2+ as a titrant. Would such a displacement of Pb2+ by Zn2+ lead to the reporting of too much or too little sulfate?
(c) In a typical analysis, a 25.00-mL sample of an industrial effluent was carried through the procedure using 50.00 mL of 0.05000 M EDTA. Titrating the excess EDTA required 12.42 mL of 0.1000 M Mg2+. Report the molar concentration of SO42– in the sample of effluent.
42. Table 9.10 provides values for the fraction of EDTA present as Y4-, αY4–. Values of αY4– are calculated using the equation
\[\alpha_\mathrm{Y^{\large 4-}}=\dfrac{[Y^{4-}]}{C_\textrm{EDTA}}\]
where [Y4−] is the concentration of the fully deprotonated EDTA and CEDTA is the total concentration of EDTA in all of its forms
\[C_\textrm{EDTA}=[\mathrm{H_6Y^{2+}}]+[\mathrm{H_5Y^+}]+[\mathrm{H_4Y}]+[\mathrm{H_3Y^-}]+[\mathrm{H_2Y^{2-}}]+[\mathrm{HY^{3-}}]+[\mathrm{Y^{4-}}]\]
Using the following equilibria
|
\(\mathrm{H_6Y^{2+}}(aq)+\mathrm{H_2O}(l)\rightleftharpoons\mathrm{H_3O^+}(aq)+\mathrm{H_5Y^+}(aq)\) |
\(K_\textrm{a1}\) |
|
\(\mathrm{H_5Y^+}(aq)+\mathrm{H_2O}(l)\rightleftharpoons\mathrm{H_3O^+}(aq)+\mathrm{H_4Y}(aq)\) |
\(K_\textrm{a2}\) |
|
\(\mathrm{H_4Y}(aq)+\mathrm{H_2O}(l)\rightleftharpoons\mathrm{H_3O^+}(aq)+\mathrm{H_3Y^-}(aq)\) |
\(K_\textrm{a3}\) |
|
\(\mathrm{H_3Y^-}(aq)+\mathrm{H_2O}(l)\rightleftharpoons\mathrm{H_3O^+}(aq)+\mathrm{H_2Y^{2-}}(aq)\) |
\(K_\textrm{a4}\) |
|
\(\mathrm{H_2Y^{2-}}(aq)+\mathrm{H_2O}(l)\rightleftharpoons\mathrm{H_3O^+}(aq)+\mathrm{HY^{3-}}(aq)\) |
\(K_\textrm{a5}\) |
|
\(\mathrm{HY^{3-}}(aq)+\mathrm{H_2O}(l)\rightleftharpoons\mathrm{H_3O^+}(aq)+\mathrm{Y^{4-}}(aq)\hspace{10mm}\) |
\(K_\textrm{a6}\) |
show that
\[\alpha_\mathrm{Y^{4-}}=\dfrac{K_\textrm{a1}K_\textrm{a2}K_\textrm{a3}K_\textrm{a4}K_\textrm{a5}K_\textrm{a6}}{d}\]
where
\[d=[\textrm H^+]^6+[\textrm H^+]^5K_\textrm{a1}+[\textrm H^+]^4K_\textrm{a1}K_\textrm{a2}+[\textrm H^+]^3K_\textrm{a1}K_\textrm{a2}K_\textrm{a3}+\]
\[[\textrm H^+]^2K_\textrm{a1}K_\textrm{a2}K_\textrm{a3}K_\textrm{a4}+[\textrm H^+]^1K_\textrm{a1}K_\textrm{a2}K_\textrm{a3}K_\textrm{a4}K_\textrm{a5}+ K_\textrm{a1}K_\textrm{a2}K_\textrm{a3}K_\textrm{a4}K_\textrm{a5}K_\textrm{a6}\]
43. Calculate or sketch titration curves for the following (unbalanced) redox titration reactions at 25oC. Assume the analyte is initially present at a concentration of 0.0100 M and that a 25.0-mL sample is taken for analysis. The titrant, which is the underlined species in each reaction, is 0.0100 M.
\((\textrm{a})\;\mathrm{V^{2+}}(aq) + \underline{\mathrm{Ce^{4+}}}(aq)\rightarrow \mathrm V^{3+}(aq) + \mathrm{Ce^3+}(aq)\)\((\textrm{b})\;\mathrm{Ti^{2+}}(aq) + \underline{\mathrm{Fe^{3+}}}(aq)\rightarrow \mathrm{Ti^{3+}}(aq) + \mathrm{Fe^{2+}}(aq)\)
\((\textrm{c})\;\mathrm{Fe^{2+}}(aq) + \underline{\ce{MnO_4^-}}(aq) \rightarrow \mathrm{Fe^{3+}}(aq) + \mathrm{Mn^{2+}}(aq)\;\textrm{(at pH = 1)}\)
44. What is the equivalence point for each titration in problem 43?
45. Suggest an appropriate indicator for each titration in problem 43.
46. The iron content of an ore can be determined by a redox titration using K2Cr2O7 as the titrant. A sample of the ore is dissolved in concentrated HCl using Sn2+ to speed its dissolution by reducing Fe3+ to Fe2+. After the sample is dissolved, Fe2+ and any excess Sn2+ are oxidized to Fe3+ and Sn4+ using MnO4–. The iron is then carefully reduced to Fe2+ by adding a 2–3 drop excess of Sn2+. A solution of HgCl2 is added and, if a white precipitate of Hg2Cl2 forms, the analysis is continued by titrating with K2Cr2O7. The sample is discarded without completing the analysis if a precipitate of Hg2Cl2 does not form, or if a gray precipitate (due to Hg) forms.
(a) Explain why the analysis is not completed if a white precipitate of Hg2Cl2 forms, or if a gray precipitate forms.
(b) Is a determinate error introduced if the analyst forgets to add Sn2+ in the step where the iron ore is dissolved?
(c) Is a determinate error introduced if the iron is not quantitatively oxidized back to Fe3+ by the MnO4–?
47. The amount of Cr3+ in an inorganic salt can be determined by a redox titration. A portion of sample containing approximately 0.25 g of Cr3+ is accurately weighed and dissolved in 50 mL of H2O. The Cr3+ is oxidized to Cr2O72– by adding 20 mL of 0.1 M AgNO3, which serves as a catalyst, and 50 mL of 10%w/v (NH4)2S2O8, which serves as the oxidizing agent. After the reaction is complete the resulting solution is boiled for 20 minutes to destroy the excess S2O82–, cooled to room temperature, and diluted to 250 mL in a volumetric flask. A 50-mL portion of the resulting solution is transferred to an Erlenmeyer flask, treated with 50 mL of a standard solution of Fe2+, and acidified with 200 mL of 1 M H2SO4, reducing the Cr2O72– to Cr3+. The excess Fe2+ is then determined by a back titration with a standard solution of K2Cr2O7 using an appropriate indicator. The results are reported as %w/w Cr3+.
(a) There are several places in the procedure where a reagent’s volume is specified (see underlined text). Which of these measurements must be made using a volumetric pipet?
(b) Excess peroxydisulfate, S2O82– is destroyed by boiling the solution. What is the effect on the reported %w/w Cr3+ if some of the S2O82– is not destroyed during this step?
(c) Solutions of Fe2+ undergo slow air oxidation to Fe3+. What is the effect on the reported %w/w Cr3+ if the standard solution of Fe2+ is inadvertently allowed to be partially oxidized?
48. The exact concentration of H2O2 in a solution that is nominally 6% w/v H2O2 can be determined by a redox titration with MnO4–. A 25-mL aliquot of the sample is transferred to a 250-mL volumetric flask and diluted to volume with distilled water. A 25-mL aliquot of the diluted sample is added to an Erlenmeyer flask, diluted with 200 mL of distilled water, and acidified with 20 mL of 25% v/v H2SO4. The resulting solution is titrated with a standard solution of KMnO4 until a faint pink color persists for 30 s. The results are reported as %w/v H2O2.
(a) Many commercially available solutions of H2O2 contain an inorganic or organic stabilizer to prevent the autodecomposition of the peroxide to H2O and O2. What effect does the presence of this stabilizer have on the reported %w/v H2O2 if it also reacts with MnO4–?
(b) Laboratory distilled water often contains traces of dissolved organic material that may react with MnO4–. Describe a simple method to correct for this potential interference.
(c) What modifications to the procedure, if any, are need if the sample has a nominal concentration of 30% w/v H2O2.
49. The amount of iron in a meteorite was determined by a redox titration using KMnO4 as the titrant. A 0.4185-g sample was dissolved in acid and the liberated Fe3+ quantitatively reduced to Fe2+ using a Walden reductor. Titrating with 0.02500 M KMnO4 requires 41.27 mL to reach the end point. Determine the %w/w Fe2O3 in the sample of meteorite.
50. Under basic conditions, MnO4– can be used as a titrant for the analysis of Mn2+, with both the analyte and the titrant forming MnO2. In the analysis of a mineral sample for manganese, a 0.5165-g sample is dissolved and the manganese reduced to Mn2+. The solution is made basic and titrated with 0.03358 M KMnO4, requiring 34.88 mL to reach the end point. Calculate the %w/w Mn in the mineral sample.
51. The amount of uranium in an ore can be determined by a redox back titration. The analysis is accomplished by dissolving the ore in sulfuric acid and reducing the resulting UO22+ to U4+ with a Walden reductor. The resulting solution is treated with an excess of Fe3+, forming Fe2+ and U6+. The Fe2+ is titrated with a standard solution of K2Cr2O7. In a typical analysis a 0.315-g sample of ore is passed through the Walden reductor and treated with 50.00 mL of 0.0125 M Fe3+. Back titrating with 0.00987 M K2Cr2O7 requires 10.52 mL. What is the %w/w U in the sample?
52. The thickness of the chromium plate on an auto fender was determined by dissolving a 30.0-cm2 section in acid, and oxidizing the liberated Cr3+ to Cr2O72– with peroxydisulfate. After removing the excess peroxydisulfate by boiling, 500.0 mg of Fe(NH4)2(SO4)2•6H2O was added, reducing the Cr2O72– to Cr3+. The excess Fe2+ was back titrated, requiring 18.29 mL of 0.00389 M K2Cr2O7 to reach the end point. Determine the average thickness of the chromium plate given that the density of Cr is 7.20 g/cm3.
53. The concentration of CO in air can be determined by passing a known volume of air through a tube containing I2O5, forming CO2 and I2. The I2 is removed from the tube by distilling it into a solution containing an excess of KI, producing I3–. The I3– is titrated with a standard solution of Na2S2O3. In a typical analysis a 4.79-L sample of air was sampled as described here, requiring 7.17 mL of 0.00329 M Na2S2O3 to reach the end point. If the air has a density of 1.23 × 10–3 g/mL, determine the parts per million CO in the air.
54. The level of dissolved oxygen in a water sample can be determined by the Winkler method. In a typical analysis a 100.0-mL sample is made basic and treated with a solution of MnSO4, resulting in the formation of MnO2. An excess of KI is added and the solution is acidified, resulting in the formation of Mn2+ and I2. The liberated I2 is titrated with a solution of 0.00870 M Na2S2O3, requiring 8.90 mL to reach the starch indicator end point. Calculate the concentration of dissolved oxygen as parts per million O2.
55. The analysis for Cl– using the Volhard method requires a back titration. A known amount of AgNO3 is added, precipitating AgCl. The unreacted Ag+ is determined by back titrating with KSCN. There is a complication, however, because AgCl is more soluble than AgSCN.
(a) Why do the relative solubilities of AgCl and AgSCN lead to a titration error?
(b) Is the resulting titration error a positive or a negative determinate error?
(c) How might you modify the procedure to prevent this eliminate this source of determinate error?
(d) Will this source of determinate error be of concern when using the Volhard method to determine Br–?
56. Voncina and co-workers suggest that a precipitation titration can be monitored by measuring pH as a function of the volume of titrant if the titrant is a weak base.14 For example, when titrating Pb2+ with CrO42– the solution containing the analyte is initially acidified to a pH of 3.50 using HNO3. Before the equivalence point the concentration of CrO42– is controlled by the solubility product of PbCrO4. After the equivalence point the concentration of CrO42– is determined by the amount of excess titrant. Considering the reactions controlling the concentration of CrO42–, sketch the expected titration curve of pH versus volume of titrant.
57. Calculate or sketch the titration curve for the titration of 50.0 mL of 0.0250 M KI with 0.0500 M AgNO3. Prepare separate titration curve using pAg and pI on the y-axis.
58. Calculate or sketch the titration curve for the titration of 25.0 mL mixture of 0.0500 M KI and 0.0500 M KSCN with 0.0500 M AgNO3.
59. A 0.5131-g sample containing KBr is dissolved in 50 mL of distilled water. Titrating with 0.04614 M AgNO3 requires 25.13 mL to reach the Mohr end point. A blank titration requires 0.65 mL to reach the same end point. Report the %w/w KBr in the sample.
60. A 0.1093-g sample of impure Na2CO3 was analyzed by the Volhard method. After adding 50.00 mL of 0.06911 M AgNO3, the sample was back titrated with 0.05781 M KSCN, requiring 27.36 mL to reach the end point. Report the purity of the Na2CO3 sample.
61. A 0.1036-g sample containing only BaCl2 and NaCl is dissolved in 50 mL of distilled water. Titrating with 0.07916 M AgNO3 requires 19.46 mL to reach the Fajans end point. Report the %w/w BaCl2 in the sample.
9.6.3 Solutions to Practice Exercises
Practice Exercise 9.1
The volume of HCl needed to reach the equivalence point is
\[V_\textrm{eq}=V_\textrm a=\dfrac{M_\textrm bV_\textrm b}{M_\textrm a}=\dfrac{\textrm{(0.125 M)(25.0 mL)}}{\textrm{0.0625 M}}=\textrm{50.0 mL}\]
Before the equivalence point, NaOH is present in excess and the pH is determined by the concentration of unreacted OH–. For example, after adding 10.0 mL of HCl
\[[\mathrm{OH^-}]=\dfrac{\textrm{(0.125 M)(25.0 mL)}-\textrm{(0.0625 M)(10.0 mL)}}{\textrm{25.0 mL + 10.0 mL}}=\textrm{0.0714 M}\]
\[[\mathrm{H_3O^+}]=\dfrac{K_\textrm w}{[\textrm{OH}^-]}=\dfrac{1.0\times10^{-14}}{\textrm{0.0714 M}}=1.40\times10^{-13}\textrm{ M}\]
the pH is 12.85.
For the titration of a strong base with a strong acid the pH at the equivalence point is 7.00.
For volumes of HCl greater than the equivalence point, the pH is determined by the concentration of excess HCl. For example, after adding 70.0 mL of titrant the concentration of HCl is
\[[\textrm{HCl}]=\dfrac{\textrm{(0.0625 M)(70.0 mL)}-\textrm{(0.125 M)(25.0 mL)}}{\textrm{70.0 mL + 25.0 mL}}=\textrm{0.0132 M}\]
giving a pH of 1.88. Some additional results are shown here.
|
Volume of HCl (mL) |
pH |
Volume of HCl (mL) |
pH |
|
0 |
13.10 |
60 |
2.13 |
|
10 |
12.85 |
70 |
1.88 |
|
20 |
12.62 |
80 |
1.75 |
|
30 |
12.36 |
90 |
1.66 |
|
40 |
11.98 |
100 |
1.60 |
|
50 |
7.00 |
Click here to return to the chapter.
Practice Exercise 9.2
The volume of HCl needed to reach the equivalence point is
\[V_\textrm{eq}=V_\textrm a=\dfrac{M_\textrm bV_\textrm b}{M_\textrm a}=\dfrac{\textrm{(0.125 M)(25.0 mL)}}{\textrm{0.0625 M}}=\textrm{50.0 mL}\]
Before adding HCl the pH is that for a solution of 0.100 M NH3.
\[K_\textrm b=\mathrm{\dfrac{[OH^-][NH_4^+]}{[NH_3]}}=\dfrac{(x)(x)}{0.125-x}=1.75\times10^{-5}\]
\[x=[\mathrm{OH^-}]=1.47\times10^{-3}\textrm{ M}\]
The pH at the beginning of the titration, therefore, is 11.17.
Before the equivalence point the pH is determined by an NH3/NH4+ buffer. For example, after adding 10.0 mL of HCl
\[[\mathrm{NH_3}]=\dfrac{\textrm{(0.125 M)(25.0 mL)}-\textrm{(0.0625 M)(10.0 mL)}}{\textrm{25.0 mL + 10.0 mL}}=\textrm{0.0714 M}\]
\[[\mathrm{NH_4^+}]=\dfrac{\textrm{(0.0625 M)(10.0 mL)}}{\textrm{25.0 mL + 10.0 mL}}=\textrm{0.0179 M}\]
\[\textrm{pH}=9.244+\log\dfrac{0.0714\textrm{ M}}{0.0179\textrm{ M}}=9.84\]
At the equivalence point the predominate ion in solution is NH4+. To calculate the pH we first determine the concentration of NH4+
\[[\mathrm{NH_4^+}]=\dfrac{\textrm{(0.125 M)(25.0 mL)}}{\textrm{25.0 mL + 50.0 mL}}=\textrm{0.0417 M}\]
and then calculate the pH
\[K_\textrm a=\mathrm{\dfrac{[H_3O^+][NH_3]}{[NH_4^+]}}=\dfrac{(x)(x)}{0.0417 - x}=5.70\times10^{-10}\]
\[x=[\mathrm{H_3O^+}]=4.88\times10^{-6}\textrm{ M}\]
obtaining a value of 5.31.
After the equivalence point, the pH is determined by the excess HCl. For example, after adding 70.0 mL of HCl
\[[\textrm{HCl}]=\dfrac{\textrm{(0.0625 M)(70.0 mL)}-\textrm{(0.125 M)(25.0 mL)}}{\textrm{25.0 mL + 70.0 mL}}=0.0132\textrm{ M}\]
and the pH is 1.88. Some additional results are shown here.
|
Volume of HCl (mL) |
pH |
Volume of HCl (mL) |
pH |
|
0 |
11.17 |
60 |
2.13 |
|
10 |
9.84 |
70 |
1.88 |
|
20 |
9.42 |
80 |
1.75 |
|
30 |
9.07 |
90 |
1.66 |
|
40 |
8.64 |
100 |
1.60 |
|
50 |
5.31 |
Click here to return to the chapter.
Practice Exercise 9.3
Figure 9.48 shows a sketch of the titration curve. The two points before the equivalence point (VHCl = 5 mL, pH = 10.24 and VHCl = 45 mL, pH = 8.24) are plotted using the pKa of 9.244 for NH4+. The two points after the equivalence point (VHCl = 60 mL, pH = 2.13 and VHCl = 80 mL, pH = 1.75 ) are from the answer to Practice Exercise 9.2.
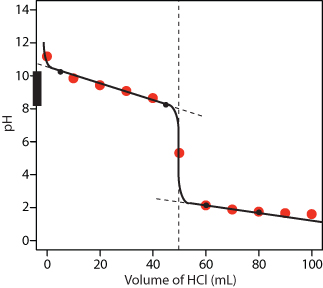
Figure 9.48 Titration curve for Practice Exercise 9.3. The black dots and curve are the approximate sketch of the titration curve. The points in red are the calculations from Practice Exercise 9.2.
Click here to return to the chapter.
Practice Exercise 9.4
Figure 9.49 shows a sketch of the titration curve. The titration curve has two equivalence points, one at 25.0 mL (H2A → HA−) and one at 50.0 mL (HA− → A2−). In sketching the curve, we plot two points before the first equivalence point using the pKa of 3 for H2A
\[\mathrm{\mathit V_{HCl} = 2.5\;mL,\;pH = 2\;and\;\mathit V_{HCl} = 22.5\;mL,\;pH = 4}\]
two points between the equivalence points using the pKa of 5 for HA–
\[\mathrm{\mathit V_{HCl} = 27.5\;mL,\;pH = 3,\;and\;\mathit V_{HCl} = 47.5\;mL,\;pH = 5}\]
and two points after the second equivalence point
\[\mathrm{\mathit V_{HCl} = 70\;mL,\;pH = 12.22\;and\;\mathit V_{HCl} = 90\;mL,\;pH = 12.46}\]
Drawing a smooth curve through these points presents us with the following dilemma—the pH appears to increase as the titrant’s volume approaches the first equivalence point and then appears to decrease as it passes through the first equivalence point. This is, of course, absurd; as we add NaOH the pH cannot decrease. Instead, we model the titration curve before the second equivalence point by drawing a straight line from the first point (VHCl = 2.5 mL, pH = 2) to the fourth (VHCl = 47.5 mL, pH = 5), ignoring the second and third points. The results is a reasonable approximation of the exact titration curve.
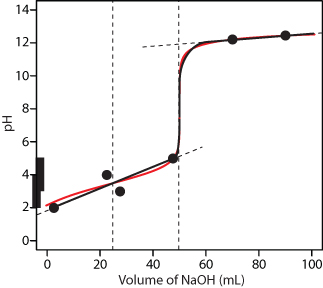
Figure 9.49 Titration curve for Practice Exercise 9.4. The black points and curve are the approximate titration curve, and the red curve is the exact titration curve.
Click here to return to the chapter.
Practice Exercise 9.5
The pH at the equivalence point is 5.31 (see Practice Exercise 9.2) and the sharp part of the titration curve extends from a pH of approximately 7 to a pH of approximately 4. Of the indicators in Table 9.4, methyl red is the best choice because its pKa value of 5.0 is closest to the equivalence point’s pH and because the pH range of 4.2–6.3 for its change in color will not produce a significant titration error.
Click here to return to the chapter.
Practice Exercise 9.6
Because salicylic acid is a diprotic weak acid, we must first determine to which equivalence point it is being titrated. Using salicylic acid’s pKa values as a guide, the pH at the first equivalence point is between a pH of 2.97 and 13.74, and the second equivalence points is at a pH greater than 13.74. From Table 9.4, phenolphthalein’s end point falls in the pH range 8.3–10.0. The titration, therefore, is to the first equivalence point for which the moles of NaOH equal the moles of salicylic acid; thus
\[\dfrac{\textrm{0.1354 mol NaOH}}{\mathrm{L}}\times0.02192\textrm{ L}=2.968\times10^{-3}\textrm{ mol NaOH}\]
\[\mathrm{2.968\times10^{-3}\;mol\;NaOH\times\dfrac{1\;mol\;C_7H_6O_3}{mol\;NaOH}\times\dfrac{138.12\;g\;C_7H_6O_3}{mol\;C_7H_6O_3}=0.4099\;g\;C_7H_6O_3}\]
\[\mathrm{\dfrac{0.4099\;g\;C_7H_6O_3}{0.4208\;g\;sample}\times100=97.41\%\;w/w\;C_7H_6O_3}\]
Because the purity of the sample is less than 99%, we reject the shipment.
Click here to return to the chapter.
Practice Exercise 9.7
The moles of HNO3 produced by pulling the air sample through the solution of H2O2 is
\[\mathrm{\dfrac{0.01012\;mol\;NaOH}{L}\times0.00914\;L\times\dfrac{1\;mol\;HNO_3}{mol\;NaOH}=9.25\times10^{-5}\;mol\;HNO_3}\]
A conservation of mass on nitrogen requires that each mole of NO2 in the sample of air produces one mole of HNO3; thus, the mass of NO2 in the sample is
\[\mathrm{9.25\times10^{-5}\;mol\;HNO_3\times\dfrac{1\;mol\;NO_2}{mol\;HNO_3}\times\dfrac{46.01\;g\;NO_2}{mol\;NO_2}=4.26\times10^{-3}\;g\;NO_2}\]
and the concentration of NO2 is
\[\mathrm{\dfrac{4.26\times10^{-3}\;g\;NO_2}{5\;L\;air}\times\dfrac{1000\;mg}{g}=0.852\;mg\;NO_2/L\;air}\]
Click here to return to the chapter.
Practice Exercise 9.8
The total moles of HCl used in this analysis is
\[\mathrm{\dfrac{1.396\;mol\;NaOH}{L}\times0.01000\;L=1.396\times10^{-2}\;mol\;HCl}\]
Of this,
\[\mathrm{\dfrac{0.1004\;mol\;NaOH}{L}\times0.03996\;L\times\dfrac{1\;mol\;HCl}{mol\;NaOH}=4.012\times10^{-3}\;mol\;HCl}\]
are consumed in the back titration with NaOH, which means that
\[\mathrm{1.396\times10^{-2}\;mol\;HCl-4.012\times10^{-3}\;mol\;HCl=9.95\times10^{-3}\;mol\;HCl}\]
react with the CaCO3. Because CO32– is dibasic, each mole of CaCO3 consumes two moles of HCl; thus
\[\mathrm{9.95\times10^{-3}\;mol\;HCl\times\dfrac{1\;mol\;CaCO_3}{2\;mol\;HCl}\times\dfrac{100.09\;g\;CaCO_3}{mol\;CaCO_3}=0.498\;g\;CaCO_3}\]
\[\mathrm{\dfrac{0.498\;g\;CaCO_3}{0.5143\;g\;sample}\times100=96.8\%\;w/w\;CaCO_3}\]
Click here to return to the chapter.
Practice Exercise 9.9
Of the two analytes, 2-methylanilinium is the stronger acid and is the first to react with the titrant. Titrating to the bromocresol purple end point, therefore, provides information about the amount of 2-methylanilinium in the sample.
\[\mathrm{\dfrac{0.200\;mol\;NaOH}{L}\times0.01965\;L\times\dfrac{1\;mol\;C_7H_{10}NCl}{mol\;NaOH}\times\dfrac{143.61\;g\;C_7H_{10}NCl}{mol\;C_7H_{10}NCl}=0.564\;g\;C_7H_{10}NCl}\]
\[\mathrm{\dfrac{0.564\;g\;C_7H_{10}NCl}{2.006\;g\;sample}\times100=28.1\%\;w/w\;C_7H_{10}NCl}\]
Titrating from the bromocresol purple end point to the phenolphthalein end point, a total of 48.41 mL – 19.65 mL, or 28.76 mL, gives the amount of NaOH reacting with 3-nitrophenol. The amount of 3-nitrophenol in the sample, therefore, is
\[\mathrm{\dfrac{0.200\;mol\;NaOH}{L}\times0.02876\;L\times\dfrac{1\;mol\;C_6H_5NO_3}{mol\;NaOH}\times\dfrac{139.11\;g\;C_6H_5NO_3}{mol\;C_6H_5NO_3}=0.800\;g\;C_6H_5NO_3}\]
\[\mathrm{\dfrac{0.800\;g\;C_6H_5NO_3}{2.006\;g\;sample}\times100=38.8\%\;w/w\;C_6H_5NO_3}\]
Click here to return to the chapter.
Practice Exercise 9.10
The first of the two visible end points is approximately 37 mL of NaOH. The analyte’s equivalent weight, therefore, is
\[\mathrm{\dfrac{0.1032\;mol\;NaOH}{L}\times0.037\;L\times\dfrac{1\;equivalent}{mol\;NaOH}=3.8\times10^{-3}\;equivalents}\]
\[EW=\mathrm{\dfrac{0.5000\;g}{3.8\times10^{-3}\;equivalents}=1.3\times10^2\;g/equivalent}\]
Click here to return to the chapter.
Practice Exercise 9.11
At ½Veq, or approximately 18.5 mL, the pH is approximately 2.2; thus, we estimate that the analyte’s pKa is 2.2.
Click here to return to the chapter.
Practice Exercise 9.12
Let’s begin with the calculations at a pH of 10. At a pH of 10 some of the EDTA is present in forms other than Y4–. To evaluate the titration curve, therefore, we need the conditional formation constant for CdY2–, which, from Table 9.11 is Kf´ = 1.1 × 1016. Note that the conditional formation constant is larger in the absence of an auxiliary complexing agent.
The titration’s equivalence point requires
\[V_\textrm{eq}=V_\textrm{EDTA}=\dfrac{M_\textrm{Cd}V_\textrm{Cd}}{M_\textrm{EDTA}}=\mathrm{\dfrac{(5.00\times10^{-3}\;M)(50.0\;mL)}{0.0100\;M}=25.0\;mL}\]
of EDTA.
Before the equivalence point, Cd2+ is present in excess and pCd is determined by the concentration of unreacted Cd2+. For example, after adding 5.00 mL of EDTA, the total concentration of Cd2+ is
\[\begin{align}
\ce{[Cd^{2+}]}&=\mathrm{\dfrac{(5.00\times10^{-3}\;M)(50.0\;mL)-(0.0100\;M)(5.00\;mL)}{50.0\;mL+5.00\;mL}}\\
&=3.64\times10^{-3}\textrm{ M}
\end{align}\]
which gives a pCd of 2.43.
At the equivalence point all the Cd2+ initially in the titrand is now present as CdY2–. The concentration of Cd2+, therefore, is determined by the dissociation of the CdY2– complex. First, we calculate the concentration of CdY2–.
\[\mathrm{[CdY^{2-}]=\dfrac{(5.00\times10^{-3}\;M)(50.0\;mL)}{50.0\;mL+25.0\;mL}=3.33\times10^{-3}\;M}\]
Next, we solve for the concentration of Cd2+ in equilibrium with CdY2–.
\[K_{\textrm{f}}'=\dfrac{\mathrm{[CdY^{2-}]}}{\mathrm{[Cd^{2+}}]C_\textrm{EDTA}}=\dfrac{3.33\times10^{-3}-x}{(x)(x)}=1.1\times10^{16}\]
Solving gives [Cd2+] as 5.50 × 10–10 M, or a pCd of 9.26 at the equivalence point.
After the equivalence point, EDTA is in excess and the concentration of Cd2+ is determined by the dissociation of the CdY2– complex. First, we calculate the concentrations of CdY2– and of unreacted EDTA. For example, after adding 30.0 mL of EDTA
\[\mathrm{[CdY^{2-}]=\dfrac{(5.00\times10^{-3}\;M)(50.0\;mL)}{50.0\;mL+30.0\;mL}=3.13\times10^{-3}\;M}\]
\[\begin{align}
C_\textrm{EDTA}&=\mathrm{\dfrac{(0.0100\;M)(30.0\;mL)-(5.00\times10^{-3}\;M)(50.0\;mL)}{50.0\;mL+30.0\;mL}}\\
&=6.25\times10^{-4}\textrm{ M}
\end{align}\]
Substituting into the equation for the conditional formation constant and solving for [Cd2+] gives
\[\mathrm{\dfrac{3.13\times10^{-3}\;M}{[Cd^{2+}](6.25\times10^{-4}\;M)}=1.1\times10^{16}}\]
[Cd2+] as 4.55× 10–16 M, or a pCd of 15.34.
The calculations at a pH of 7 are identical, except the conditional formation constant for CdY2– is 1.5 × 1013 instead of 1.1 × 1016. The following table summarizes results for these two titrations as well as the results from Table 9.13 for the titration of Cd2+ at a pH of 10 in the presence of 0.0100 M NH3 as an auxiliary complexing agent.
|
Volume of |
pCd |
pCd at pH 10 w/ |
pCd |
|
0 |
2.30 |
3.36 |
2.30 |
|
5.00 |
2.43 |
3.49 |
2.43 |
|
10.0 |
2.60 |
3.66 |
2.60 |
|
15.0 |
2.81 |
3.87 |
2.81 |
|
20.0 |
3.15 |
4.20 |
3.15 |
|
23.0 |
3.56 |
4.62 |
3.56 |
|
25.0 |
9.26 |
9.77 |
7.83 |
|
27.0 |
14.94 |
14.95 |
12.08 |
|
30.0 |
15.34 |
15.33 |
12.48 |
|
35.0 |
15.61 |
15.61 |
12.78 |
|
40.0 |
15.76 |
15.76 |
12.95 |
|
45.0 |
15.86 |
15.86 |
13.08 |
|
50.0 |
15.94 |
15.94 |
13.18 |
Examining these results allows us to draw several conclusions. First, in the absence of an auxiliary complexing agent the titration curve before the equivalence point is independent of pH (compare columns 2 and 4). Second, for any pH, the titration curve after the equivalence point is the same regardless of whether or not an auxiliary complexing agent is present (compare columns 2 and 3). Third, the largest change in pH through the equivalence point occurs at higher pHs and in the absence of an auxiliary complexing agent. For example, from 23.0 mL to 27.0 mL of EDTA the change in pCd is 11.38 at a pH of 10, 10.33 at a pH of 10 and in the presence of 0.0100 M NH3, and 8.52 at a pH of 7.
Click here to return to the chapter.
Practice Exercise 9.13
Figure 9.50 shows a sketch of the titration curves. The two points before the equivalence point (VEDTA = 5 mL, pCd = 2.43 and VEDTA = 15 mL, pCd = 2.81) are the same for both pHs and are taken from the results of Practice Exercise 9.12. The two points after the equivalence point for a pH of 7 (VEDTA = 27.5 mL, pCd = 12.2 and VEDTA = 50 mL, pCd = 13.2 ) are plotted using the logKf´ of 13.2 for CdY2-. The two points after the equivalence point for a pH of 10 (VEDTA = 27.5 mL, pCd = 15.0 and VEDTA = 50 mL, pCd = 16.0) are plotted using the logKf´ of 16.0 for CdY2-.
Figure 9.50 Titration curve for Practice Exercise 9.13. The black dots and curve are the approximate sketches of the two titration curves. The points in red are the calculations from Practice Exercise 9.12 for a pH of 10, and the points in green are the calculations from Practice Exercise 9.12 for a pH of 7.
Click here to return to the chapter.
Practice Exercise 9.14
In an analysis for hardness we treat the sample as if Ca2+ is the only metal ion reacting with EDTA. The grams of Ca2+ in the sample, therefore, is
\[\mathrm{\dfrac{0.0109\;mol\;EDTA}{L}\times0.02363\;L\times\dfrac{1\;mol\;Ca^{2+}}{mol\;EDTA}=2.58\times10^{-4}\;mol\;Ca^{2+}}\]
\[\mathrm{2.58\times10^{-4}\;mol\;Ca^{2+}\times\dfrac{1\;mol\;CaCO_3}{mol\;Ca^{2+}}\times\dfrac{100.09\;g\;CaCO_3}{mol\;CaCO_3}=0.0258\;g\;CaCO_3}\]
and the sample’s hardness is
\[\mathrm{\dfrac{0.0258\;g\;CaCO_3}{0.1000\;L}\times\dfrac{1000\;mg}{g}=258\;mg\;CaCO_3/L}\]
Click here to return to the chapter.
Practice Exercise 9.15
The titration of CN– with Ag+ produces a metal-ligand complex of Ag(CN)22–; thus, each mole of AgNO3 reacts with two moles of NaCN. The grams of NaCN in the sample is
\[\mathrm{\dfrac{0.1018\;mol\;AgNO_3}{L}\times0.03968\;L\times\dfrac{2\;mol\;NaCN}{mol\;AgNO_3}\times\dfrac{49.01\;g\;NaCN}{mol\;NaCN}=0.3959\;g\;NaCN}\]
and the purity of the sample is
\[\mathrm{\dfrac{0.3959\;g\;NaCN}{0.4482\;g\;sample}\times100=88.33\%\;w/w\;NaCN}\]
Click here to return to the chapter.
Practice Exercise 9.16
The total moles of EDTA used in this analysis is
\[\mathrm{\dfrac{0.02011\;mol\;EDTA}{L}\times0.02500\;L = 5.028\times10^{-4}\;mol\;EDTA}\]
Of this,
\[\mathrm{\dfrac{0.01113\;mol\;Mg^{2+}}{L}\times0.00423\;L\times\dfrac{1\;mol\;EDTA}{mol\;Mg^{2+}}=4.708\times10^{-5}\;mol\;EDTA}\]
are consumed in the back titration with Mg2+, which means that
\[\mathrm{5.028\times10^{-4}\;mol\;EDTA-4.708\times10^{-5}\;mol\;EDTA=4.557\times10^{-4}\;mol\;EDTA}\]
react with the BaSO4. Each mole of BaSO4 reacts with one mole of EDTA; thus
\[\mathrm{4.557\times10^{-4}\;mol\;EDTA\times\dfrac{1\;mol\;BaSO_4}{mol\;EDTA}\times\dfrac{1\;mol\;Na_2SO_4}{mol\;BaSO_4}\times\dfrac{142.04\;g\;Na_2SO_4}{mol\;Na_2SO_4}=0.06473\;g\;Na_2SO_4}\]
\[\mathrm{\dfrac{0.06473\;g\;Na_2SO_4}{0.1557\;g\;sample}\times100=41.23\%\;w/w\;Na_2SO_4}\]
Click here to return to the chapter.
Practice Exercise 9.17
The volume of Tl3+ needed to reach the equivalence point is
\[V_\textrm{eq}=V_\textrm{Tl}=\dfrac{M_\textrm{Sn}V_\textrm{Sn}}{M_\textrm{Tl}}=\mathrm{\dfrac{(0.050\;M)(50.0\;mL)}{0.100\;M}=25.0\;mL}\]
Before the equivalence point, the concentration of unreacted Sn2+ and the concentration of Sn4+ are easy to calculate. For this reason we find the potential using the Nernst equation for the Sn4+/Sn2+ half-reaction.For example, the concentrations of Sn2+ and Sn4+ after adding 10.0 mL of titrant are
\[\mathrm{[Sn^{2+}]=\dfrac{(0.050\;M)(50.0\;mL)-(0.100\;M)(10.0\;mL)}{50.0\;mL+10.0\;mL}=0.0250\;M}\]
\[\mathrm{[Sn^{4+}]=\dfrac{(0.100\;M)(10.0\;mL)}{50.0\;mL+10.0\;mL}=0.0167\;M}\]
and the potential is
\[E=\mathrm{+0.139\;V-\dfrac{0.05916}{2}\log\dfrac{0.0250\;M}{0.0167\;M}=+0.134\;V}\]
After the equivalence point, the concentration of Tl+ and the concentration of excess Tl3+ are easy to calculate. For this reason we find the potential using the Nernst equation for the Tl3+/Tl+ half-reaction. For example, after adding 40.0 mL of titrant, the concentrations of Tl+ and Tl3+ are
\[\mathrm{[Tl^+]=\dfrac{(0.0500\;M)(50.0\;mL)}{50.0\;mL+40.0\;mL}=0.0278\;M}\]
\[\mathrm{[Tl^{3+}]=\dfrac{(0.100\;M)(40.0\;mL)-(0.0500\;M)(50.0\;mL)}{50.0\;mL+40.0\;mL}=0.0167\;M}\]
and the potential is
\[E=\mathrm{+0.77\;V-\dfrac{0.05916}{2}\log\dfrac{0.0278\;M}{0.0167\;M}=+0.76\;V}\]
At the titration’s equivalence point, the potential, Eeq, potential is
\[E_\textrm{eq}=\mathrm{\dfrac{0.139\;V+0.77\;V}{2}=0.45\;V}\]
Some additional results are shown here.
|
Volume of Tl3+ (mL) |
E (V) |
Volume of Tl3+ (mL) |
E (V) |
|
5 |
0.121 |
30 |
0.75 |
|
10 |
0.134 |
35 |
0.75 |
|
15 |
0.144 |
40 |
0.76 |
|
20 |
0.157 |
45 |
0.76 |
|
25 |
0.45 |
50 |
0.76 |
Click here to return to the chapter.
Practice Exercise 9.18
Figure 9.51 shows a sketch of the titration curve. The two points before the equivalence point
\[\mathrm{\mathit V_{Tl} = 2.5\;mL,\;\mathit E = +0.109\;V\;and\;\mathit V_{Tl} = 22.5\; mL,\;\mathit E = +0.169\;V}\]
are plotted using the redox buffer for Sn4+/Sn2+, which spans a potential range of +0.139 ± 0.5916/2. The two points after the equivalence point
\[\mathrm{\mathit V_{Tl} = 27.5\;mL,\;\mathit E = +0.74\;V\;and\;\mathit V_{EDTA} = 50\;mL,\;\mathit E = +0.77\;V}\]
are plotted using the redox buffer for Tl3+/Tl+, which spans the potential range of +0.139 ± 0.5916/2.

Figure 9.51 Titration curve for Practice Exercise 9.18. The black dots and curve are the approximate sketch of the titration curve. The points in red are the calculations from Practice Exercise 9.17.
Click here to return to the chapter.
Practice Exercise 9.19
The two half reactions are
\[\textrm{Ce}^{4+}(aq)+e^-\rightarrow \textrm{Ce}^{3+}(aq)\]
\[\mathrm{U^{4+}}(aq)+\mathrm{2H_2O}\rightarrow \mathrm{UO_2^{2+}}(aq)+\mathrm{4H^+}(aq)+2e^-\]
for which the Nernst equations are
\[E=E^{\circ}\mathrm{_{Ce^{4+}/Ce^{3+}}-\dfrac{0.05916}{1}\log\dfrac{[Ce^{3+}]}{[Ce^{4+}]}}\]
\[E=E^{\circ}\mathrm{_{UO_2^{2+}/U^{4+}}-\dfrac{0.05916}{2}\log\dfrac{[U^{4+}]}{[UO_2^{2+}][H^+]^4}}\]
Before adding these two equations together we must multiply the second equation by 2 so that we can combine the log terms; thus
\[3E=E^{\circ}\mathrm{_{Ce^{4+}/Ce^{3+}}}+2E^{\circ}\mathrm{_{UO_2^{2+}/U^{4+}}-0.05916\log\dfrac{[Ce^{3+}][U^{4+}]}{[Ce^{4+}][UO_2^{2+}][H^+]^4}}\]
At the equivalence point we know that
\[\mathrm{[Ce^{3+}]=2\times[UO_2^{2+}]}\]
\[\mathrm{[Ce^{4+}]=2\times[U^{4+}]}\]
Substituting these equalities into the previous equation and rearranging gives us a general equation for the potential at the equivalence point.
\[3E=E^{\circ}\mathrm{_{Ce^{4+}/Ce^{3+}}}+2E^{\circ}\mathrm{_{UO_2^{2+}/U^{4+}}-0.05916\log\dfrac{2[UO_2^{2+}][U^{4+}]}{2[U^{4+}][UO_2^{2+}][H^+]^4}}\]
\[E=\dfrac{E^{\circ}_\mathrm{Ce^{4+}/Ce^{3+}}+2E^{\circ}_\mathrm{UO_2^{2+}/U^{4+}}}{3}-\dfrac{0.05916}{3}\log\dfrac{1}{[\textrm H^+]^4}\]
\[E=\dfrac{E^{\circ}_\mathrm{Ce^{4+}/Ce^{3+}}+2E^{\circ}_\mathrm{UO_2^{2+}/U^{4+}}}{3}+\dfrac{0.05916\times4}{3}\log[\textrm H^+]\]
\[E=\dfrac{E^{\circ}_\mathrm{Ce^{4+}/Ce^{3+}}+2E^{\circ}_\mathrm{UO_2^{2+}/U^{4+}}}{3}-0.07888\textrm{ pH}\]
At a pH of 1 the equivalence point has a potential of
\[E_\textrm{eq}=\dfrac{1.72 + 2\times0.327}{3}-0.07888\times1=0.712\textrm{ V}\]
Click here to return to the chapter.
Practice Exercise 9.20
Because we have not been provided with a balanced reaction, let’s use a conservation of electrons to deduce the stoichiometry. Oxidizing C2O42–, in which each carbon has a +3 oxidation state, to CO2, in which carbon has an oxidation state of +4, requires one electron per carbon, or a total of two electrons for each mole of C2O42–. Reducing MnO4–, in which each manganese is in the +7 oxidation state, to Mn2+ requires five electrons. A conservation of electrons for the titration, therefore, requires that two moles of KMnO4 (10 moles of e-) reacts with five moles of Na2C2O4 (10 moles of e-).
The moles of KMnO4 used in reaching the end point is
\[\mathrm{(0.0400\;M\;KMnO_4)\times(0.03562\;L\;KMnO_4)=1.42\times10^{-3}\;mol\;KMnO_4}\]
which means that the sample contains
\[\mathrm{1.42\times10^{-3}\;mol\;KMnO_4\times\dfrac{5\;mol\;Na_2C_2O_4}{2\;mol\;KMnO_4}=3.55\times10^{-3}\;mol\;Na_2C_2O_4}\]
Thus, the %w/w Na2C2O4 in the sample of ore is
\[\mathrm{3.55\times10^{-3}\;mol\;Na_2C_2O_4\times\dfrac{134.00\;g\;Na_2C_2O_4}{mol\;Na_2C_2O_4}=0.476\;g\;Na_2C_2O_4}\]
\[\mathrm{\dfrac{0.476\;g\;Na_2C_2O_4}{0.5116\;g\;sample}\times100=93.0\%\;w/w\;Na_2C_2O_4}\]
Click here to return to the chapter.
Practice Exercise 9.21
For a back titration we need to determine the stoichiometry between Cr2O72– and the analyte, C2H6O, and between Cr2O72– and the titrant, Fe2+. In oxidizing ethanol to acetic acid, the oxidation state of carbon changes from –2 in C2H6O to 0 in C2H4O2. Each carbon releases two electrons, or a total of four electrons per C2H6O. In reducing Cr2O72–, in which each chromium has an oxidation state of +6, to Cr3+, each chromium loses three electrons, for a total of six electrons per Cr2O72–. Oxidation of Fe2+ to Fe3+ requires one electron. A conservation of electrons requires that each mole of K2Cr2O7 (6 moles of e-) reacts with six moles of Fe2+ (6 moles of e-), and that four moles of K2Cr2O7 (24 moles of e-) react with six moles of C2H6O (24 moles of e-).
The total moles of K2Cr2O7 reacting with C2H6O and with Fe2+ is
\[\mathrm{(0.0200\;M\;K_2Cr_2O_7)\times(0.05000\;L\;\ce{I_3^-})=1.00\times10^{-3}\;mol\;K_2Cr_2O_7}\]
The back titration with Fe2+ consumes
\[\mathrm{0.02148\;L\;Fe^{2+}\times\dfrac{0.1014\;mol\;Fe^{2+}}{L\;Fe^{2+}}\times\dfrac{1\;mol\;K_2Cr_2O_7}{6\;mol\;Fe^{2+}}=3.63\times10^{-4}\;mol\;K_2Cr_2O_7}\]
Subtracting the moles of K2Cr2O7 reacting with Fe2+ from the total moles of K2Cr2O7 gives the moles reacting with the analyte.
\[\mathrm{1.00\times10^{-3}\;K_2Cr_2O_7-3.63\times10^{-4}\;mol\;K_2Cr_2O_7=6.37\times10^{-4}\;mol\;K_2Cr_2O_7}\]
The grams of ethanol in the 10.00-mL sample of diluted brandy is
\[\mathrm{6.37\times10^{-4}\;mol\;K_2Cr_2O_7\times\dfrac{6\;mol\;C_2H_6O}{4\;mol\;K_2Cr_2O_7}\times\dfrac{46.50\;g\;C_2H_6O}{mol\;C_2H_6O}=0.0444\;g\;C_2H_6O}\]
The %w/v C2H6O in the brandy is
\[\mathrm{\dfrac{0.0444\;g\;C_2H_6O}{10.00\;mL\;dilute\;brandy}\times\dfrac{500.0\;mL\;dilute\;brandy}{5.00\;mL\;brandy}\times100=44.4\%\;w/v\;C_2H_6O}\]
Click here to return to the chapter.
Practice Exercise 9.22
The first task is to calculate the volume of NaCl needed to reach the equivalence point; thus
\[V_\textrm{eq}=V_\textrm{NaCl}=\dfrac{M_\textrm{Ag}V_\textrm{Ag}}{M_\textrm{NaCl}}=\mathrm{\dfrac{(0.0500\;M)(50.0\;mL)}{(0.100\;M)}=25.0\;mL}\]
Before the equivalence point the titrand, Ag+, is in excess. The concentration of unreacted Ag+ after adding 10.0 mL of NaCl, for example, is
\[\mathrm{[Ag^+]=\dfrac{(0.0500\;M)(50.0\;mL)-(0.100\;M)(10.0\;mL)}{50.0\;mL+10.0\;mL}=2.50\times10^{-2}\;M}\]
which corresponds to a pAg of 1.60. To find the concentration of Cl– we use the Ksp for AgCl; thus
\[[\mathrm{Cl^-}]=\dfrac{K_\textrm{sp}}{[\mathrm{Ag^+}]}=\dfrac{1.8\times10^{-10}}{2.50\times10^{-2}}=7.2\times10^{-9}\textrm{ M}\]
or a pCl of 8.14.
At the titration’s equivalence point, we know that the concentrations of Ag+ and Cl– are equal. To calculate their concentrations we use the Ksp expression for AgCl; thus
\[K_\textrm{sp}=\mathrm{[Ag^+][Cl^-]}=(x)(x)=1.8\times10^{-10}\]
Solving for x gives a concentration of Ag+ and the concentration of Cl– as 1.3 × 10–5 M, or a pAg and a pCl of 4.89.
After the equivalence point, the titrant is in excess. For example, after adding 35.0 mL of titrant
\[\mathrm{[Cl^-]=\dfrac{(0.100\;M)(35.0\;mL)-(0.0500\;M)(50.0\;mL)}{50.0\;mL+35.0\;mL}=1.18\times10^{-2}\;M}\]
or a pCl of 1.93. To find the concentration of Ag+ we use the Ksp for AgCl; thus
\[[\mathrm{Ag^+}]=\dfrac{K_\textrm{sp}}{[\mathrm{Cl^-}]}=\dfrac{1.8\times10^{-10}}{1.18\times10^{-2}}=1.5\times10^{-8}\textrm{ M}\]
or a pAg of 7.82. The following table summarizes additional results for this titration.
|
Volume of NaCl (mL) |
pAg |
pCl |
|
0 |
1.30 |
– |
|
5.00 |
1.44 |
8.31 |
|
10.0 |
1.60 |
8.14 |
|
15.0 |
1.81 |
7.93 |
|
20.0 |
2.15 |
7.60 |
|
25.0 |
4.89 |
4.89 |
|
30.0 |
7.54 |
2.20 |
|
35.0 |
7.82 |
1.93 |
|
40.0 |
7.97 |
1.78 |
|
45.0 |
8.07 |
1.68 |
|
50.0 |
8.14 |
1.60 |
Click here to return to the chapter.
Practice Exercise 9.23
The titration uses
\[\mathrm{\dfrac{0.1078\;M\;KSCN}{L}\times0.02719\;L=2.931\times10^{-3}\;mol\;KSCN}\]
The stoichiometry between SCN– and Ag+ is 1:1; thus, there are
\[\mathrm{2.931\times10^{-3}\;mol\;Ag^+\times\dfrac{107.87\;g\;Ag}{mol\;Ag}=0.3162\;g\;Ag}\]
in the 25.00 mL sample. Because this represents ¼ of the total solution, there are 0.3162 × 4 or 1.265 g Ag in the alloy. The %w/w Ag in the alloy is
\[\mathrm{\dfrac{1.265\;g\;Ag}{1.963\;g\;sample}\times100=64.44\%\;w/w\;Ag}\]
Click here to return to the chapter.