
11.5: Most Solutions are Not Ideal
- Page ID
- 204501
If we plot the partial pressure of one component, \(P_1\), above a mixture with a mole fraction \(x_1\), we should get a straight line with a slope of \(P^*_1\) (Raoult's law). Above non-ideal solutions the graph will no longer be a straight line but a curve. However towards \(x_1=1\) the curve typically approaches the Raoult line. On the other extreme, there often is a more or less linear region as well, but with a different slope (Figure 24.5.1 ). This means that we can identify two limiting laws:
- For \(x \rightarrow 0\): Henry's law:
\[P_1 = K_H x_1 \nonumber \]
- For \(x \rightarrow 1\): Raoult's law:
\[P_1 = P^*_1 x_1 \nonumber \]
This implies that the straight line that indicates the Henry expression will intersect the y-axis at \(x=1\) (pure compound) at a different point than \(P^*\). For \(x \rightarrow 0\) (low concentrations) we can speak of component 1 being the solute (the minority component). At the other end \(x \rightarrow 1\) it plays the role of the solvent (majority component).
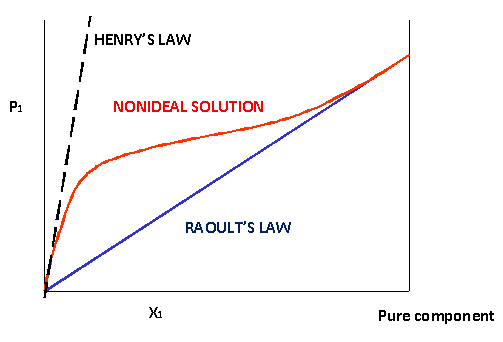
Another thing to note is that \(P^*\) is a property of one pure component, the value of \(K_H\) by contrast is a property of the combination of two components, so it needs to be measured for each solute-solvent combination.
As you can see we have a description for both the high and the low end, but not in the middle. In general, the more modest the deviations from ideality the larger the range of validity of the two limiting laws. The way to determine \(K_H\) would be to actually determine vapor pressures. How about the other component? Do we need to measure them too? Fortunately we can use thermodynamics to answer this question with no. There is a handy expression that saves us the trouble.