
8.4: Infrared (IR) Spectroscopy
- Page ID
- 239580
Introduction
Photon energies associated with the infrared (from 1 to 15 kcal/mole) are not large enough to excite electrons, but may induce vibrational excitation of covalently bonded atoms and groups.
The covalent bonds in molecules are not rigid sticks or rods, such as found in molecular model kits, but are more like stiff springs that can be stretched and bent. The mobile nature of organic molecules was noted in the chapter concerning conformational isomers. We must now recognize that, in addition to the facile rotation of groups about single bonds, molecules experience a wide variety of vibrational motions, characteristic of their component atoms. Consequently, virtually all organic compounds will absorb infrared radiation that corresponds in energy to these vibrations. Infrared spectrometers, similar in principle to the UV-Visible spectrometer described elsewhere, permit chemists to obtain absorption spectra of compounds that are a unique reflection of their molecular structure.
Vibrational Spectroscopy
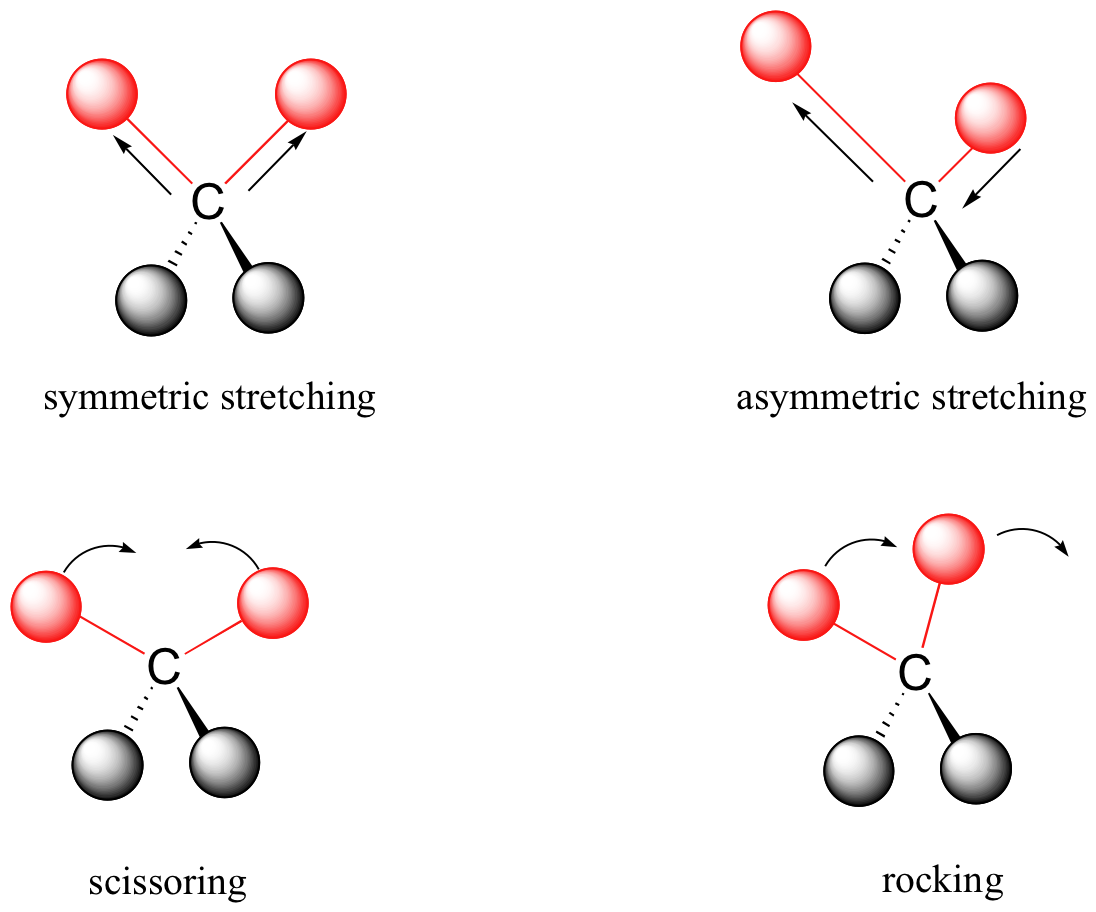
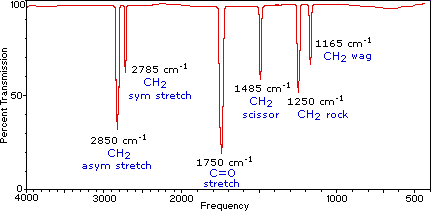
A molecule composed of n-atoms has 3n degrees of freedom, six of which are translations and rotations of the molecule itself. This leaves 3n-6 degrees of vibrational freedom (3n-5 if the molecule is linear). Vibrational modes are often given descriptive names, such as stretching, bending, scissoring, rocking and twisting. The four-atom molecule of formaldehyde, the gas phase spectrum of which is shown below, provides an example of these terms. If a ball & stick model of formaldehyde is not displayed to the right of the spectrum, press the view ball&stick model button on the right. We expect six fundamental vibrations (12 minus 6), and these have been assigned to the spectrum absorptions. To see the formaldehyde molecule display a vibration, click one of the buttons under the spectrum, or click on one of the absorption peaks in the spectrum.
Covalent bonds in organic molecules are not rigid sticks – rather, they behave more like springs. At room temperature, organic molecules are always in motion, as their bonds stretch, bend, and twist. These complex vibrations can be broken down mathematically into individual vibrational modes, a few of which are illustrated below.
The energy of molecular vibration is quantized rather than continuous, meaning that a molecule can only stretch and bend at certain 'allowed' frequencies. If a molecule is exposed to electromagnetic radiation that matches the frequency of one of its vibrational modes, it will in most cases absorb energy from the radiation and jump to a higher vibrational energy state - what this means is that the amplitude of the vibration will increase, but the vibrational frequency will remain the same. The difference in energy between the two vibrational states is equal to the energy associated with the wavelength of radiation that was absorbed. It turns out that it is the infrared region of the electromagnetic spectrum which contains frequencies corresponding to the vibrational frequencies of organic bonds.
An IR Spectrum
We will use a ketone sample to illustrate this process. The sample is irradiated with infrared light and the carbonyl bond will specifically absorb light with this same frequency, which by equations 4.1 and 4.2 corresponds to a wavelength of 5.83 x 10-6 m and an energy of 4.91 kcal/mol. When the carbonyl bond absorbs this energy, it jumps up to an excited vibrational state.
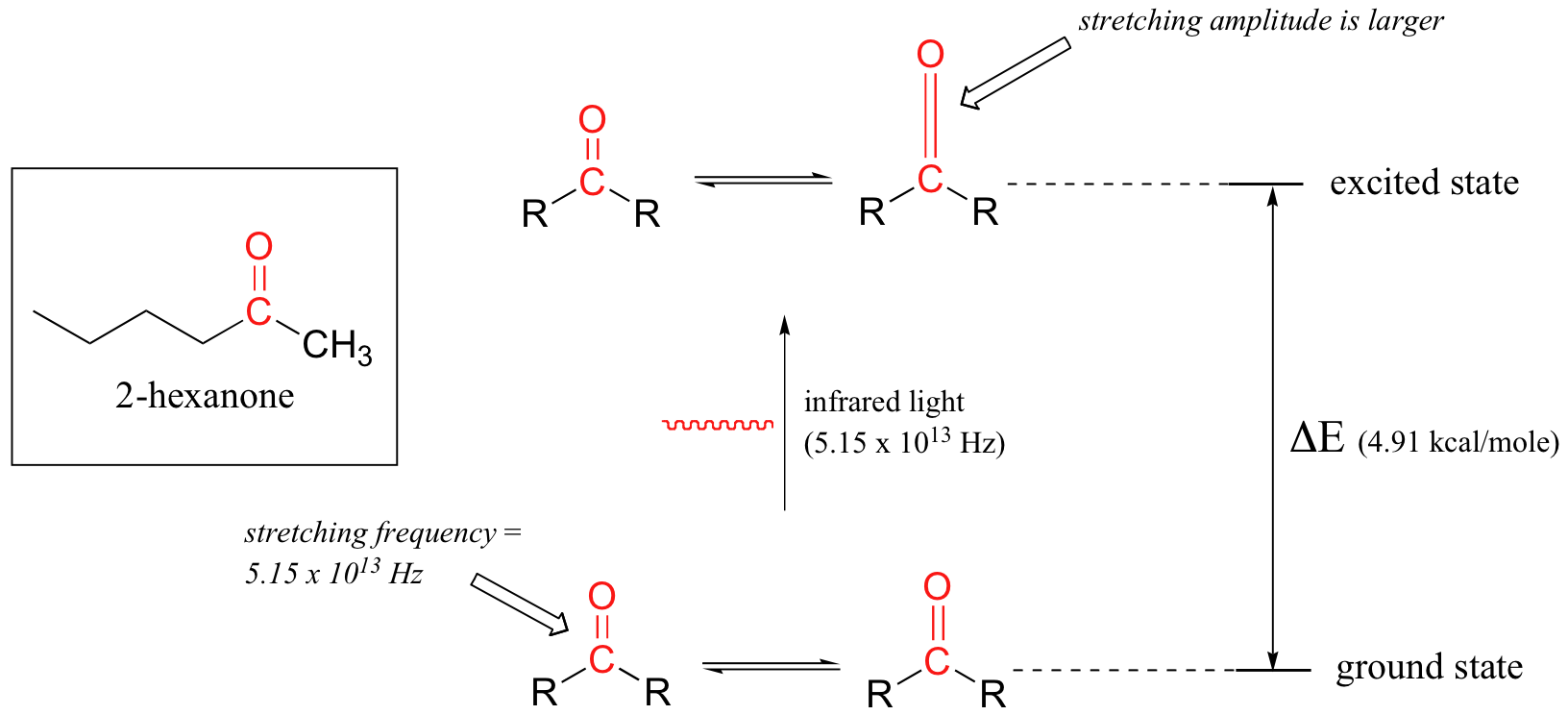
The value of ΔE - the energy difference between the low energy (ground) and high energy (excited) vibrational states - is equal to 4.91 kcal/mol, the same as the energy associated with the absorbed light frequency. The molecule does not remain in its excited vibrational state for very long, but quickly releases energy to the surrounding environment in form of heat, and returns to the ground state.
With an instrument called an infrared spectrophotometer, we can 'see' this vibrational transition. In the spectrophotometer, infrared light with frequencies ranging from about 1013 to 1014 Hz is passed though our sample of cyclohexane. Most frequencies pass right through the sample and are recorded by a detector on the other side.
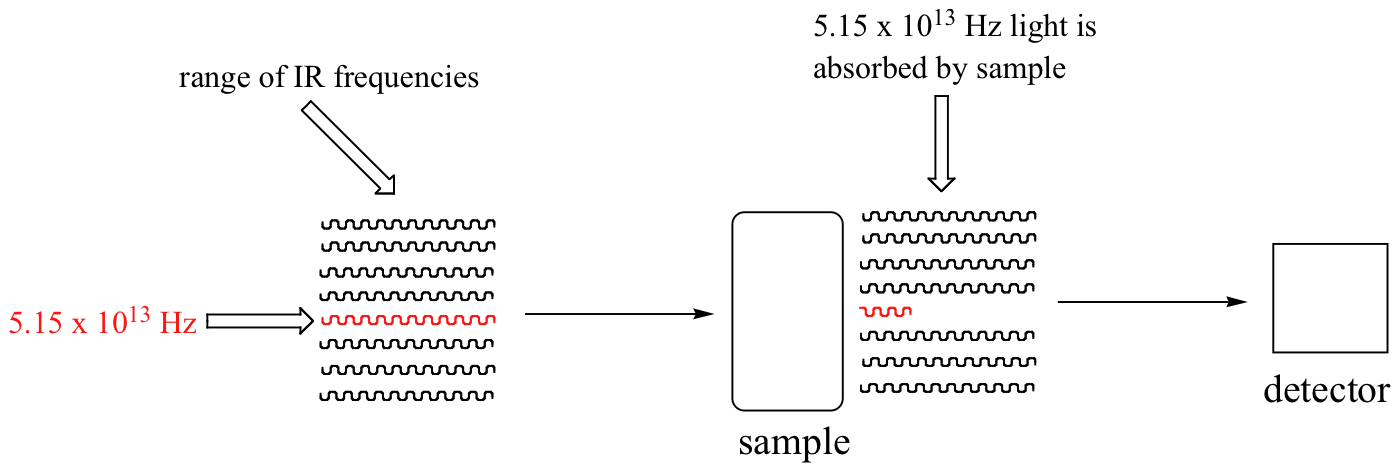
Our 5.15 x 1013 Hz carbonyl stretching frequency, however, is absorbed by the 2-hexanone sample, and so the detector records that the intensity of this frequency, after having passed through the sample, is something less than 100% of its initial intensity.
The vibrations of a 2-hexanone molecule are not, of course, limited to the simple stretching of the carbonyl bond. The various carbon-carbon bonds also stretch and bend, as do the carbon-hydrogen bonds, and all of these vibrational modes also absorb different frequencies of infrared light.
The power of infrared spectroscopy arises from the observation that different functional groups have different characteristic absorption frequencies. The carbonyl bond in a ketone, as we saw with our 2-hexanone example, typically absorbs in the range of 5.11 - 5.18 x 1013 Hz, depending on the molecule. The carbon-carbon triple bond of an alkyne, on the other hand, absorbs in the range 6.30 - 6.80 x 1013 Hz. The technique is therefore very useful as a means of identifying which functional groups are present in a molecule of interest. If we pass infrared light through an unknown sample and find that it absorbs in the carbonyl frequency range but not in the alkyne range, we can infer that the molecule contains a carbonyl group but not an alkyne.
Now, let's look at some actual output from IR spectroscopy experiments. Below is the IR spectrum for 2-hexanone.
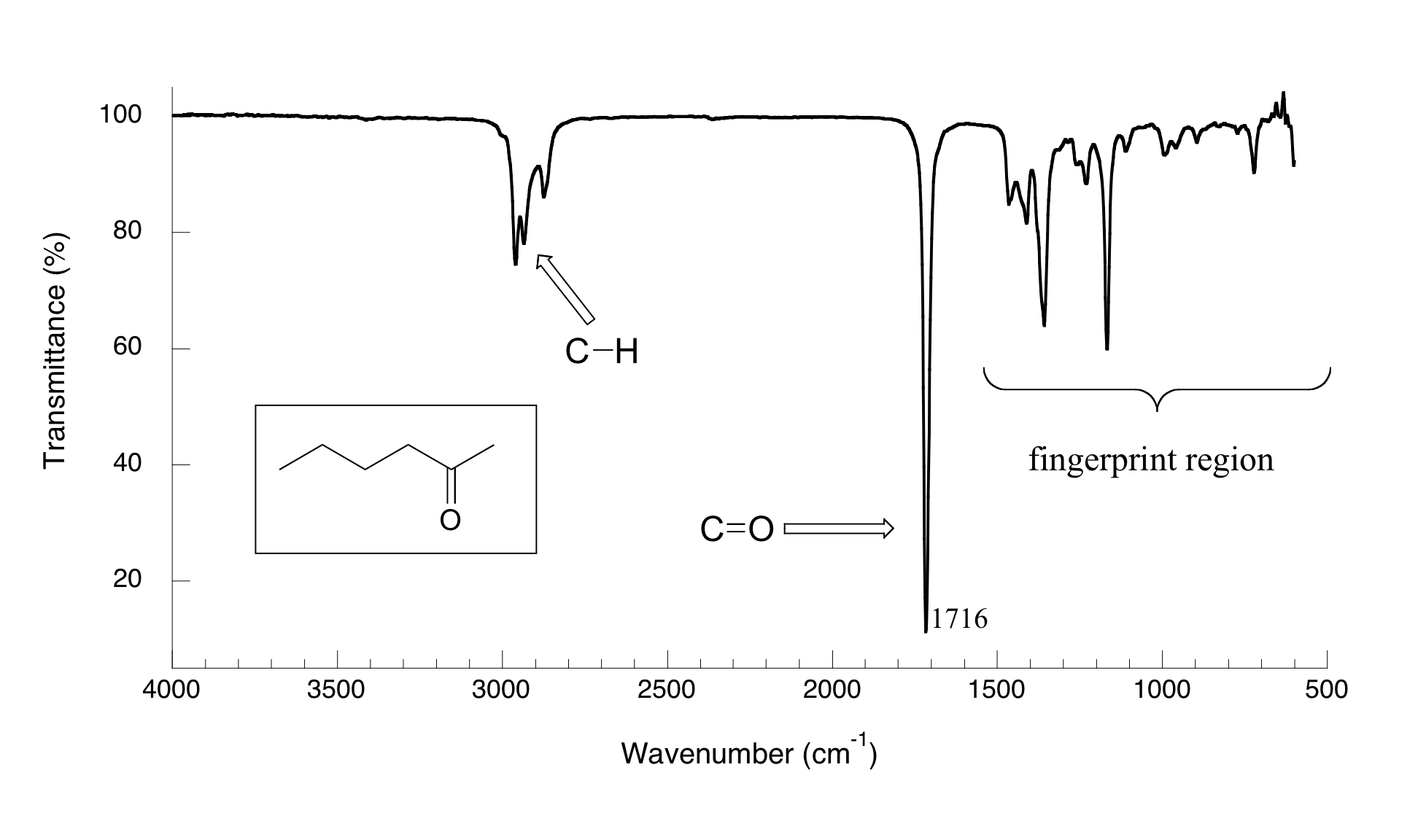
There are a number of things that need to be explained in order for you to understand what it is that we are looking at. On the horizontal axis we see IR wavelengths expressed in terms of a unit called wavenumber (cm-1), which tells us how many waves fit into one centimeter. On the vertical axis we see ‘% transmittance’, which tells us how strongly light was absorbed at each frequency (100% transmittance means no absorption occurred at that frequency). The solid line traces the values of % transmittance for every wavelength – the ‘peaks’ (which are actually pointing down) show regions of strong absorption. For some reason, it is typical in IR spectroscopy to report wavenumber values rather than wavelength (in meters) or frequency (in Hz). The ‘upside down’ vertical axis, with absorbance peaks pointing down rather than up, is also a curious convention in IR spectroscopy. We wouldn’t want to make things too easy for you!
A calculator for interconverting these frequency and wavelength values is provided on the right. Simply enter the value to be converted in the appropriate box, press "Calculate" and the equivalent number will appear in the empty box.
Infrared spectra may be obtained from samples in all phases (liquid, solid and gaseous). Liquids are usually examined as a thin film sandwiched between two polished salt plates (note that glass absorbs infrared radiation, whereas NaCl is transparent). If solvents are used to dissolve solids, care must be taken to avoid obscuring important spectral regions by solvent absorption. Perchlorinated solvents such as carbon tetrachloride, chloroform and tetrachloroethene are commonly used. Alternatively, solids may either be incorporated in a thin KBr disk, prepared under high pressure, or mixed with a little non-volatile liquid and ground to a paste (or mull) that is smeared between salt plates.
Frequency - Wavelength Converter |
|
|---|---|
| Frequency in cm-1 Wavelength in μ |
|
Gas Phase Infrared Spectrum of Formaldehyde, H2C=O |
||
|---|---|---|
 |
||
|
Ball&Stick Model Spacefill Model Stick Model Motion Off |
|
The exact frequency at which a given vibration occurs is determined by the strengths of the bonds involved and the mass of the component atoms. For a more detailed discussion of these factors Click Here. In practice, infrared spectra do not normally display separate absorption signals for each of the 3n-6 fundamental vibrational modes of a molecule. The number of observed absorptions may be increased by additive and subtractive interactions leading to combination tones and overtones of the fundamental vibrations, in much the same way that sound vibrations from a musical instrument interact. Furthermore, the number of observed absorptions may be decreased by molecular symmetry, spectrometer limitations, and spectroscopic selection rules. One selection rule that influences the intensity of infrared absorptions, is that a change in dipole moment should occur for a vibration to absorb infrared energy. Absorption bands associated with C=O bond stretching are usually very strong because a large change in the dipole takes place in that mode.
Some General Trends:
- Stretching frequencies are higher than corresponding bending frequencies. (It is easier to bend a bond than to stretch or compress it.)
- Bonds to hydrogen have higher stretching frequencies than those to heavier atoms.
- Triple bonds have higher stretching frequencies than corresponding double bonds, which in turn have higher frequencies than single bonds.(Except for bonds to hydrogen).
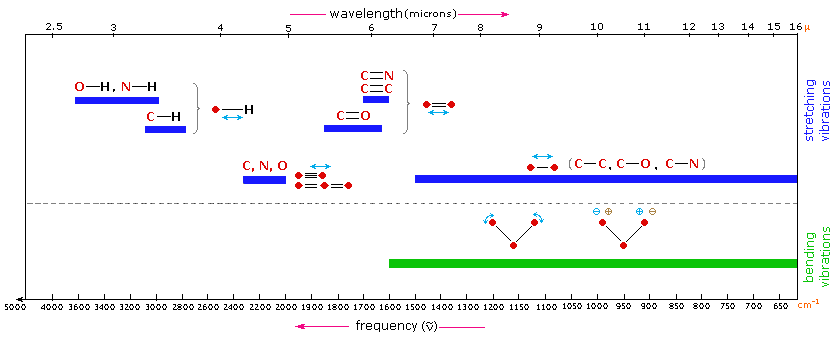
The general regions of the infrared spectrum in which various kinds of vibrational bands are observed are outlined in the following chart. Note that the blue colored sections above the dashed line refer to stretching vibrations, and the green colored band below the line encompasses bending vibrations. The complexity of infrared spectra in the 1450 to 600 cm-1 region makes it difficult to assign all the absorption bands, and because of the unique patterns found there, it is often called the fingerprint region. Absorption bands in the 4000 to 1450 cm-1 region are usually due to stretching vibrations of diatomic units, and this is sometimes called the group frequency region.
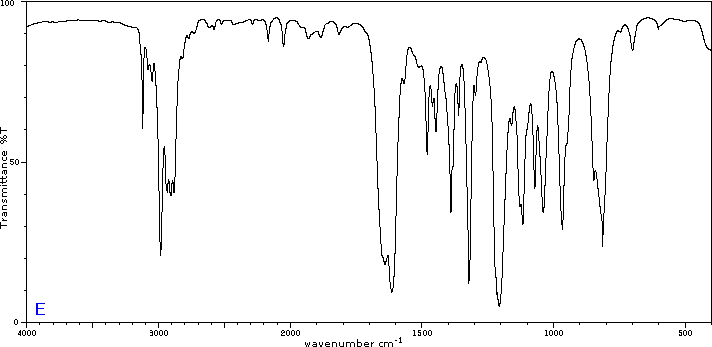
To illustrate the usefulness of infrared absorption spectra, examples for five C4H8O isomers are presented below their corresponding structural formulas. Try to associate each spectrum (A - E) with one of the isomers in the row above it.
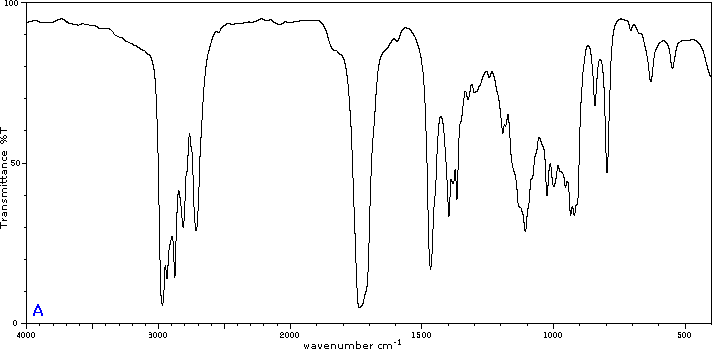
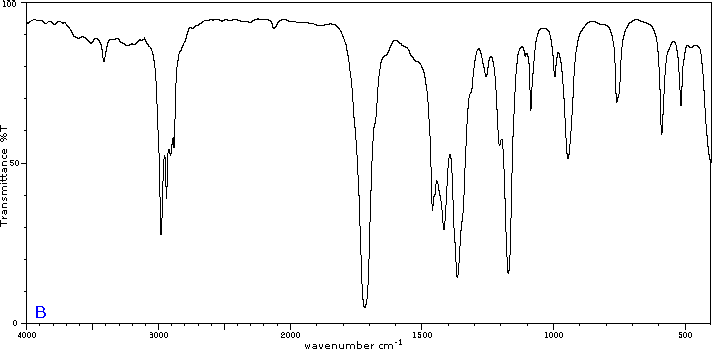
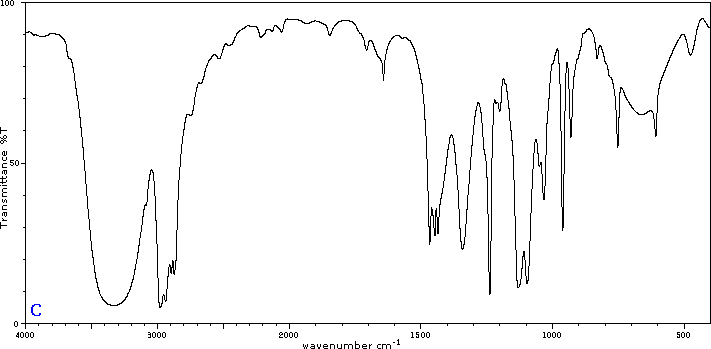
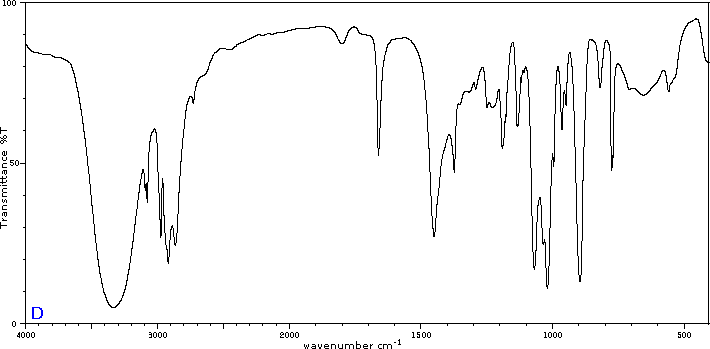
Internal Links
Contributors
- William Reusch, Professor Emeritus (Michigan State U.), Virtual Textbook of Organic Chemistry
Contributors and Attributions
Dr. Dietmar Kennepohl FCIC (Professor of Chemistry, Athabasca University)
Prof. Steven Farmer (Sonoma State University)
William Reusch, Professor Emeritus (Michigan State U.), Virtual Textbook of Organic Chemistry
Organic Chemistry With a Biological Emphasis by Tim Soderberg (University of Minnesota, Morris)