
3.6: MALDI-TOF
- Page ID
- 379451
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)Proteins and peptides have been characterized by high pressure liquid chromatography (HPLC) or SDS PAGE by generating peptide maps. These peptide maps have been used as fingerprints of protein or as a tool to know the purity of a known protein in a known sample. Mass spectrometry gives a peptide map when proteins are digested with amino end specific, carboxy end specific, or amino acid specific digestive enzymes. This peptide map can be used to search a sequence database to find a good match from the existing database. This is because the more accurately the peptide masses are known, the less chance there is of bad matches.
Introduction
Electron spray ionization coupled to triple quadrupole (TSQ) and ion trap mass spectrometers (ITMS) and matrix assisted laser desorption ionization (MALDI) coupled to time of flight (TOF) analyzers have been successful for obtaining very accurate mass measurements. TOF, TSQ, and ITMS can give mass accuracies better than 0.1. MALDI-TOF mass spectra (MS) is a good tool for screening peptide masses of tryptic digests. This method is more effective because it requires relatively less intense sample preparation since the matrix is less susceptible to interferences caused by salts and detergents. Secondly MALDI-TOF-MS generates peptides containing only one charge and show only one peak in spectrum which facilitates data interpretation.
Schematic and Theory of MALDI
MALDI is a very sensitive technique for determining the mass of proteins, peptides, or polymers. Protein masses are identity of proteins and thus help in proteomics. Thus MALDI allows protein identification. MALDI sample preparation is relatively fast and easy. It is a first choice when it comes to protein study. Proteins, peptides, and polymers are fragile and tend to fragment when ionized by other ionization techniques.
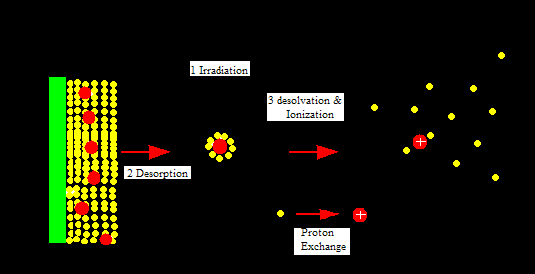
MALDI is attached to a time of flight (TOF) analyzer which measures time it takes for the molecules to travel a fixed distance. MALDI is a soft ionization technique in which a short laser pulse, instead of continuous laser, of nitrogen gas usually around 237 nm is used to ionize molecules. A protein or peptide sample is placed on a target plate and mixed with an appropriate matrix on the target plate. The mixture of sample and matrix crystallizes due to the vacuum environment and then is irradiated with a short laser pulse. The sample molecules and the matrix now enter gas phase. This leads to release of matrix, samples molecules, and ions from the target plate. The ions then accelerate in TOF analyzer because they are subject to equal electric field. TOF is a field-free flight tube. The ions travel in a strait and linear direction to the detector. The mass to charge ration (m/z) of the sample ions can be calculated using the equation T= C1(m/z)0.5 +C2. C1 and C2 are instrumental constants which can be determined with compounds of known mass. This equation is derived from the fact that potential energy equals kinetic energy.
\[KE = \dfrac{1}{2}mv^2 \tag{1}\]
\[v = \sqrt{\dfrac{2KE}{m}} \tag{2}\]
Since velocity is distance/time \(v=d/t\) substitute it in equation 2 and solve for \(t\) to get eq. 3
t = m1/2 * d/(2KE)1/2 eq.3
The distance the molecules travel and their kinetic energy is constant. So it is replaced by C1. Furthermore since the relationship between the t and m1/2 is linear an intercept of C2 is added to get equation of a line.
t = C1(m/z)1/2 + C2 eq.4
Sample preparation
Biomolecules such as proteins, peptides, sugars, and large organic molecules such as polymers, dendrimers and other macro molecules can be analyzed using MALDI. Sample preparation for MALDI is very simple; however, it is one of the most crucial steps in the MALDI analysis process. MALDI is more tolerant to sample contaminants, but contaminants can seriously disturb incorporation of sample molecules with growing matrix crystals. This results in bad spots on the target plate, leading to low signal to noise ratio, resolution, and sensitivity.
Samples can be prepared in two different ways. One removes the contaminants before applying them on to the matrix and one removes the contaminants after the sample is spotted on to the target plate either before or after adding the matrix.
Miniaturized chromatographic set-up is used for the first approach, while some scientists have cleaned in-gel digest of proteins using reverse phase(RP) HPLC microcolumns packed with different types of RP-HPLC beads (1). Now tips are also packed with RP or ion-exchange resin to remove salts and detergents from protein mixtures, and their effectiveness is shown by their recent commercialization. Tips and columns effectively remove MALDI contaminants and give small volume of sample, this can result in possible highly sensitive MALDI analysis of the samples.
Purification of biological samples on target plates involves synthetic membranes or surfaces. Membranes go on top of the target plate and the biological samples are spotted on top of the membranes. Biological samples interact with the membrane through strong hydrophobic forces. This enables samples to remain on the membrane while the buffers and salts are washed away. Then the MALDI-matrix solution is added to the purified samples on the target plate, ready for analysis. Perfluorosulfonated ionomer films, polyethylene membranes, nonporous polyurethane membranes, and C8 and C18 extraction disks are examples of membranes that have been used successfully in the past for biological mixtures. Self-assembled monolayers (SAMs) of octadecyl mercaptan on gold-sputtered disposable MALDI probe tips have been used to concentrate the sample and to act as a purification device. However, it is unfeasible because it requires overnight sample incubation to fully concentrate at the probe tip.
Matrix
The function of the matrix is adsorborption of energy from laser pulse, and then transfer to sample“thereby causing desorption of the analyte molecules in an expanding plume, to ionize the desorbed analyte molecules and to prevent aggregation of the analyte molecules”(2). The matrix molecules for MALDI are chosen on the basis on fulfillment of requirement that matrix molecules must be able to absorb ultraviolet wavelength of usually 237nm, low volatility and ability to transfer protons to the sample molecules. For proteins samples typical MALDI matrix consist of cinnamic acid and hydroxylated benzoic acid derivatives.
2,5-dihydroxybenzoic acid is more tolerant to the sample contaminants because it excludes them during crystallization process(3). The use of specially prepared thin matrix layers uses fast evaporation setup, which not only improves the sensitivity and resolution, but also allows the samples to be extensively washed, removing salts and detergents. Since the sensitivity depends on the concentration of the sample on the target plate, samples can be concentrated using PR-HPLC or bead-peptide concentration. In bead-peptide concentration RP-chromatographic beads are added to the proteins or peptide samples, and these samples preferentially bind to the beads through hydrophobic interactions while the contaminants like salts and chaotrophes do not. After a short incubation, the peptide-bead solution is harvested using pellets from a centrifugation, and dried in speed vacuum concentrator. In both cases, highly concentrated pellets of peptide-bound beads are obtained, which can be transferred to MALDI and left to dry. Because the beads are hydrophobic in nature, they form a cluster in highly concentrated spot (<1mm^2) on MALDI target plate after drying. Peptides elute on the target plate by a small volume of aqueous/organic MALDI-matrix solution and become incorporated into the growing matrix crystals at the same time. This process allows 10 to 100 fmol to be enough to be loaded on to the gels.
Sensitivity
Sensitivity of MALDI depends on sample preparation and the preparation of sample/matrix layer. They must be optimized by trial and error according to sample size, type, and previous history. Preparing a very thin matrix layer and applying of sample so that the sample is on the outer layer of matrix, give a sensitivity level of low attomole (10^-18). This method makes it possible for “…removal of salts present in the analyte solution by a sample washing procedure away from the sample very simply” (4). Small matrix spots using nanoliter volumes of matrix and analyte solution, combined purification, concentration, and application procedures have also given similar sensitivities (5). In this method, matrix is adsorbed to nanoliter bed volume reversed-phase column prepared in an Eppendorf GeLoder tip. Then the column is washed and the sample is eluted with a few nanoliter volume of matrix onto the target plate.
Sensitivity is reduced with increasing molecular weight. The sensitivity is two to three magnitudes lower for proteins than it is for peptides. So the sensitivity of proteins is in the femtomole range.
Structural Information
Structural information of proteins can be determined by digesting proteins with specific endoprotease like trypsin, AspN, and GluC. MALDI is one of the best spectrometric techniques for direct analysis of peptide mixtures. Signals of peptides are suppressed because there is a competition for charge or optimal position in the matrix. Therefore signal intensity does not necessarily reflect the quantities of different peptides in the mixtures. Complete sequences can be obtained from a combination of spectra recorded in different modes, like positive and negative, matrices, and different enzyme digestion.
Sequence information is also possible to get from PSD. This is possible by controlling the voltage of the reflector, which results in different m/z ranges on the detector and generates a PSD spectrum. A large sample amount is required since only a small fraction goes under PSD. Additionally, the fragmentation can not be controlled since different site of a peptide can get fragmented. This makes it very had to get complete sequences of a peptide. Alternatively, collision cells are included to the flight tube in MALDI-TOF by some manufacturers, to have controlled fragmentation by collision-induced dissociation.
Large amounts of in-source fragmentation occurs before initiation of the acceleration voltage called in-source decay in delayed extraction equipped MALDI-TOF, which only yield long regions of sequence-specific ions (6). C-terminal sequence ladders can be generated by digestion of peptides with carboxypepsidase and N-terminal sequence ladders can be obtained by Edman degradation using low percentage of phenyltiocarbamate rather than phenyoisothiocarbamate in the coupling reaction. These ladders in the mixtures of peptides can be an alternative to sequence-specific fragment ions. This process often gives a lot of sequence information.
Secondary protein modifications can also be determined using MALDI-TOF-MS. The steps involved in determining secondary modifications are measuring mass of the intact protein, knowing the protein’s primary sequence, and generating site-specific information by direct mass spectrometric peptide mapping of a mixture derived by proteolytic cleavage of the proteins. In tandem (TOF/TOF) configurations, MALDI instruments can provide protein sequence data, as well.
In tandem mass spectrometry, an ion of a particular mass is selected (that's the first stage of the analysis) and fragmented. Its constituent fragment ions are then mass-analyzed a second time (that's the tandem stage) to reveal data about the molecule's structure or sequence; single-stage TOF instruments lack this capability (though some fragmentation does occur via "post-source decay" as the ions traverse the flight tube).
Some companies offer tandem MALDI instruments based on hybrid mass analyzer configurations. Applied Biosystems' QSTAR, for instance, couples an optional MALDI source with a quadrupole-time-of-flight mass analyzer, as does Waters Corporation's MALDI Q-Tof Premier.
TOF analyzer
Modes
Ions can travel in a linear fashion and be detected by the detector at the opposite end as an ion source. This is called linear TOF. It is different from a reflectron TOF, in which ions are reflected to electrostatic mirror and detected by another detector. Linear TOF spectrum is limited in resolution leading to low mass accuracy. This is because initially different amount of kinetic energy can be attained by the ions with the same charge. This leads to different (m/z) ratio of ions which have different initial velocities. This is partially corrected by reflectron TOF. High energy ions penetrate deeper into the reflectron, taking longer distance and time, while low energy ions do not penetrate as deep into the reflectron and take a shorter path and time. This leads to correction of different times of ions with the same mass and charge. This leads to an increase of resolution to 10,000.
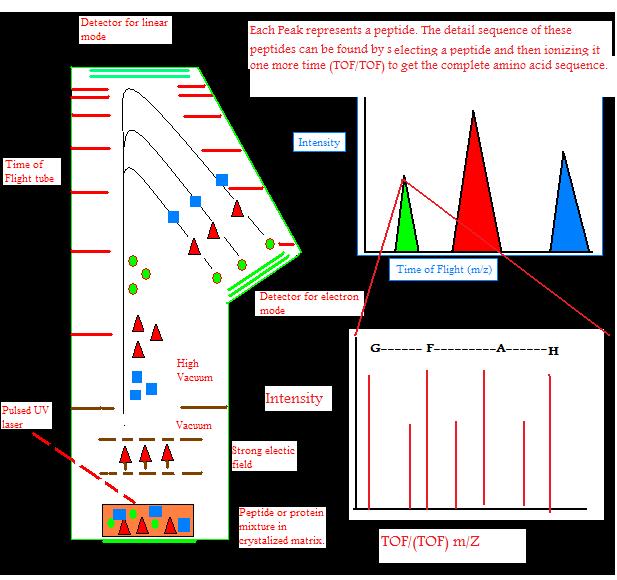
The amount of energy put into ions by the laser initially can also be corrected by a technique called delayed extraction (DE) in which acceleration voltage is applied slightly after the laser pulse. DE TOF increases resolution to 2000.
RE TOF can give full isotopic resolution for molecules up to 15 kDa. However 12C-only ion peak will have very low intensity for molecules bigger than 5kDa. Resolution is further decreased because each ion decays after acceleration and its time of flight can not be adjusted by RE TOF because decay has already occurred. These ions are not detected in RE TOF. However, these ions can be detected in linear TOF, but resolution still decreases because linear TOF has no way of correcting different energy inputs to an ion with the same mass and charge. It is better to use linear mode to get spectra and determine isotopically averaged mass for molecules bigger than 5-10 kDa. Resolution depends on the size of the molecules in a sample. The greater the size that the sample molecule has, the lower the resolution of it.
MALDI-TOF can only be compared to ESI because they are two ways of directly analyzing proteins, peptides, and polymers. MALDI-TOF samples can be reanalyzed while ESI samples can not because ESI is connected to LC column, and the analysis is limited to the width of the chromatographic peak. MALDI-TOF can scan 10 spectras for a peak 10 seconds wide per second, while it takes ESI almost 15 second. MALDI-TOF-MS generates mostly ions +/- 1 charge while ESI generates a charge for every 8-10 amino acids. That means ESI spectra are considerably more complex than MALDI spectra. Coulombic repulsion increases as the charge increase, leading to data deviation, and in MALDI spectra, this repulsion does not occur. However, multiple charges in ESI give better resolution because the higher the mass to charge ratio, the harder it is to get good resolution. In general MALDI is faster than ESI, and enables higher throughput. But ESI is more sensitive.
References
- Proteomics in functional genomics : protein structure analysis / edited by P. Jollès and H. Jörnva
- Fey SJ et al. (1997) Proteome analysis of Saccharomyces cerevisiae: A methodological outline. Electrophoresis 18: 1361-1372 2. Karas M. et al. (1998) Laser desorption ionization of proteins with molecular mass exceeding 10,000 daltons. Anal Chem 60: 2299-2301
- Patterson SD, Aebersold R (1995) Mass spectrometric approaches for the identification of gel-separated proteins. Electrophoresis 16: 1791-1814
- Vorm O. Roepstroff P, Mann M (1994) Improved resolution and very high sensitivity in MALDI-TOF of matrix surfaces made by fast evaporation. Anal Chem 66: 3281-3287
- Jespersen S, Niessen WM, et al. (1994) Attomole detection of proteins by matrix assisted laser desorption/ionization mass spectrometry with the use of picoliter vials. Rapid Comm. Mass Spectrom 8: 581-584
- Reiber DC, Grower TA, et al. (1998) Identifying proteins and matrix-assisted laser desorption/ionization in source fragmentation data combined with databaseresearching. Anal Chem 70: 673-678
Contributors
- Basir Syed