
10.5: The \(\pi\)-Electron Approximation of Conjugation
- Page ID
- 63714
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)- Demonstrate how Hückel's theory approximates the full molecular orbital picture of molecules by treating the \(\sigma\)-bonding and \(\pi\)-bonding networks independently.
Molecular orbital theory has been very successfully applied to large conjugated systems, especially those containing chains of carbon atoms with alternating single and double bonds. An approximation introduced by Hückel in 1931 considers only the delocalized p electrons moving in a framework of \(\pi\)-bonds. This is, in fact, a more sophisticated version of a free-electron model.
The simplest hydrocarbon to consider that exhibits \(\pi\) bonding is ethylene (ethene), which is made up of four hydrogen atoms and two carbon atoms. Experimentally, we know that the H–C–H and H–C–C angles in ethylene are approximately 120°. This angle suggests that the carbon atoms are sp2 hybridized, which means that a singly occupied sp2 orbital on one carbon overlaps with a singly occupied s orbital on each H and a singly occupied sp2 lobe on the other C. Thus each carbon forms a set of three \(\sigma\) bonds: two C–H (sp2 + s) and one C–C (sp2 + sp2) (part (a) of Figure 10.5.1 ).
.svg?revision=1&size=bestfit&width=811&height=286)
The Hückel approximation is used to determine the energies and shapes of the \(\pi\) molecular orbitals in conjugated systems. Within the Hückel approximation, the covalent bonding in these hydrocarbones can be separated into two independent "frameworks": the \(\sigma\)-bonding framework and the the \(\sigma\)-bonding framework. The wavefunctions used to describe the bonding orbitals in each framework results from different combinations of atomic orbitals. The method limits itself to addressing conjugated hydrocarbons and specifically only \(\pi\) electron molecular orbitals are included because these determine the general properties of these molecules; the sigma electrons are ignored. This is referred to as sigma-pi separability and is justified by the orthogonality of \(\sigma\) and \(\pi\) orbitals in planar molecules. For this reason, the Hückel method is limited to planar systems. Hückel approximation assumes that the electrons in the \(\pi\) bonds “feel” an electrostatic potential due to the entire \(\sigma\)-bonding framework in the molecule (i.e. it focuses only on the formation of \(\pi\) bonds, given that the \(\sigma\) bonding framework has already been formed).
A conjugated system has a region of overlapping p-orbitals, bridging the interjacent single bonds, that allow a delocalization of \(\pi\) electrons across all the adjacent aligned p-orbitals. These \(\pi\) electrons do not belong to a single bond or atom, but rather to a group of atoms.
Ethylene
Before considering the Hückel treatment for ethylene, it is beneficial to review the general bonding picture of the molecule. Bonding in ethylene involves the \(sp^2\) hybridization of the \(2s\), \(2p_x\), and \(2p_y\) atomic orbitals on each carbon atom; leaving the \(2p_z\) orbitals untouched (Figure 10.5.2 ).

The use of hybrid orbitals in the molecular orbital approach describe here is merely a convenience and not invoking valence bond theory (directly). An identical description can be extracted using exclusively atomic orbitals on carbon, but the interpretation of the resulting wavefunctions is less intuitive. For example, the ith molecular orbital can be described via hybrid orbitals
\[ | \psi_1\rangle = c_1 | sp^2_1 \rangle + c_2 | 1s_a \rangle \nonumber \nonumber \]
or via atomic orbitals.
\[ | \psi_1\rangle = a_1 | 2s \rangle + a_1 | 2p_x \rangle + a_1 | 2p_y \rangle + a_4| 1s_a \rangle \nonumber \nonumber \]
where \(\{a_i\}\) and \(\{c_i\}\) are coefficients of the expansion. Either describe will work and both are identical approaches since
\[| sp^2_1 \rangle = b_1 | 2s \rangle + b_1 | 2p_x \rangle + b_1 | 2p_y \rangle \nonumber \nonumber \]
where \(\{c_i\}\) are coefficients describing the hybridized orbital.
The bonding occurs via the mixing of the electrons in the \(sp^2\) hybrid orbitals on carbon and the electrons in the \(1s\) atomic orbitals of the four hydrogen atoms (Figure 10.5.1 ; left) resulting in the \(\sigma\)-bonding framework. The \(\pi\)-bonding framework results from the unhybridized \(2p_z\) orbitals (Figure 10.5.2 ; right). The independence of these two frameworks is demonstrated in the resulting molecular orbital diagram in Figure 10.5.3 ; Hückel theory is concerned only with describing the molecular orbitals and energies of the \(\pi\) bonding framework.
.svg?revision=1&size=bestfit&width=529&height=320)
Hückel treatment is concerned only with describing the molecular orbitals and energies of the \(\pi\) bonding framework.
Since Hückel theory is a special consideration of molecular orbital theory, the molecular orbitals \(| \psi_i \rangle\) can be described as a linear combination of the \(2p_z\) atomic orbitals \(\phi\) at carbon with their corresponding \(\{c_i\}\) coefficients:
\[ | \psi_i \rangle =c_1 | \phi_{1} \rangle +c_2 | \phi_2 \rangle \label{LCAO} \]
This equation is substituted in the Schrödinger equation:
\[ \hat{H} | \psi_i \rangle =E_i | \psi_i \rangle \nonumber \]
with \(\hat{H}\) the Hamiltonian and \(E_i\) the energy corresponding to the molecular orbital to give:
\[ \hat{H} c_{1} | \phi _{1} \rangle +\hat{H} c_{2} | \phi _{2} \rangle =E c_{1} | \phi _{1} \rangle +E c_{2} | \phi _{2} \rangle \label{SEq} \]
If Equation \(\ref{SEq}\) is multiplied by \(\langle \phi _{1}| \) (and integrated), then
\[c_1(H_{11} - ES_{11}) + c_2(H_{12} - ES_{12}) = 0 \label{Eq1} \]
where \( H_{ij}\) are the Hamiltonian matrix elements (see note below)
\[ H_{ij} = \langle \phi_i | \hat{H} | \phi_j \rangle = \int \phi _{i}H\phi _{j}\mathrm {d} v\nonumber \]
and \( S_{ij} \) are the overlap integrals.
\[ S_{ij}= \langle \phi_i | \phi_j \rangle = \int \phi _{i}\phi _{j}\mathrm {d} v\nonumber \]
If Equation \(\ref{SEq}\) is multiplied by \( \langle \phi _{2} | \) (and integrated), then
\[c_1(H_{21} - ES_{21}) + c_2(H_{22} - ES_{22}) = 0 \label{Eq2} \]
Both Equations \(\ref{Eq1}\) and \(\ref{Eq2}\) can better represented in matrix notation,
\[ {\begin{bmatrix}c_{1}(H_{11}-ES_{11})+c_{2}(H_{12}-ES_{12})\\c_{1}(H_{21}-ES_{21})+c_{2}(H_{22}-ES_{22})\\\end{bmatrix}}=0\nonumber \]
or more simply as a product of matrices.
\[\begin{bmatrix} H_{11} - ES_{11} & H_{12} - ES_{12} \\ H_{21} - ES_{21} & H_{22} - ES_{22} \\ \end{bmatrix} \times \begin{bmatrix} c_1 \\ c_2 \\ \end{bmatrix}= 0 \label{master} \]
All diagonal Hamiltonian integrals \( H_{ii}\) are called Coulomb integrals and those of type \(H_{ij}\) are called resonance integrals. Both integrals are negative and the resonance integrals determines the strength of the bonding interactions. The equations described by Equation \(\ref{master}\) are called the secular equations and will also have the trivial solution of
\[ c_1 = c_2 = 0 \nonumber \]
Within linear algebra, the secular equations in Equation \(\ref{master}\) will also have a non-trivial solution, if and only if, the secular determinant is zero
\[ \left| \begin{array} {cc} H_{11} - ES_{11} & H_{12} - ES_{12} \\ H_{21} - ES_{21} & H_{22} - ES_{22} \\ \end{array}\right| = 0 \label{SecDet} \]
or in shorthand notation
\[ \text{det}(H -ES) =0\nonumber \]
Everything in Equation \(\ref{SecDet}\) is a known number except \(E\). Since the secular determinant for ethylene is a \(2 \times 2\) matrix, finding \(E\), requires solving a quadratic equation (after expanding the determinant)
\[ ( H_{11} - ES_{11} ) ( H_{22} - ES_{22} ) - ( H_{21} - ES_{21} )( H_{12} - ES_{12} ) = 0\nonumber \]
There will be two values of \(E\) which satisfy this equation and they are the molecular orbital energies. For ethylene, one will be the bonding energy and the other the antibonding energy for the \(\pi\)-orbitals formed by the combination of the two carbon \(2p_z\) orbitals (Equation \(\ref{LCAO}\)). However, if more than two \(| \phi \rangle\) atomic orbitals were used, e.g., in a bigger molecule, then more energies would be estimated by solving the secular determinant.
Solving the secular determinant is simplified within Hückel method via the following four assumptions:
- All overlap integrals \(S_{ij}\) are set equal to zero. This is quite reasonable since the \(\pi-\) orbitals are directed perpendicular to the direction of their bonds (Figure 10.5.1 ). This assumption is often call neglect of differential overlap (NDO).
- All resonance integrals \(H_{ij}\) between non-neighboring atoms are set equal to zero.
- All resonance integrals \(H_{ij}\) between neighboring atoms are equal and set to \(\beta\).
- All coulomb integrals \(H_{ii}\) are set equal to \(\alpha\).
These assumptions are mathematically expressed as
\[ H_{11}=H_{22}=\alpha\nonumber \]
\[ H_{12}=H_{21}=\beta\nonumber \]
Assumptions 1 means that the overlap integral between the two atomic orbitals is 0
\[ S_{11}=S_{22}=1\nonumber \]
\[ S_{12}=S_{21}=0\nonumber \]
The Coulomb integrals
\[H_{ii}= \langle \phi _i|H| \phi _i \rangle \nonumber \nonumber \]
and resonance integrals.
\[H_{ij}= \langle \phi _i|H| \phi _j \rangle \,\,\, (i \neq i) \nonumber \nonumber \]
are often described within the matrix representation of the Hamiltonian (specifically within the \( | \phi \rangle\) basis):
\[ \hat{H} = \begin{bmatrix} H_{11} & H_{12} \\ H_{21} & H_{22} \end {bmatrix} \nonumber \nonumber \]
or within the Hückel assumptions
\[ \hat{H} = \begin{bmatrix} \alpha & \beta \\ \beta & \alpha \end {bmatrix} \nonumber \nonumber \]
The Hückel assumptions reduces Equation \(\ref{master}\) in two homogeneous equations:
\[\begin{bmatrix} \alpha - E & \beta \\ \beta & \alpha - E \\ \end{bmatrix} \times \begin{bmatrix} c_1 \\ c_2 \\ \end{bmatrix}= 0 \label{Eq12} \]
if Equation \(\ref{Eq12}\) is divided by \(\beta\):
\[\begin{bmatrix} \dfrac{\alpha - E}{\beta} & 1 \\ 1 & \dfrac{\alpha - E}{\beta} \\ \end{bmatrix} \times \begin{bmatrix} c_1 \\ c_2 \\ \end{bmatrix}= 0\nonumber \]
and then a new variable \(x\) is defined
\[ x = \dfrac {\alpha -E}{\beta} \label{new} \]
then Equation \(\ref{Eq12}\) simplifies to
\[\begin{bmatrix} x & 1 \\ 1 & x \\ \end{bmatrix} \times \begin{bmatrix} c_1 \\ c_2 \\ \end{bmatrix}= 0 \label{seceq} \]
The trivial solution gives both wavefunction coefficients equal to zero and the other (non-trivial) solution is determined by solving the secular determinant
\[ \begin{vmatrix}x&1\\1&x\\\end{vmatrix}=0\nonumber \]
which when expanded is \(x^{2}-1=0\) so \( x=\pm 1\).
Knowing that \(E=\alpha -x\beta \) from Equation \(\ref{new}\), the energy levels can be found to be
\[ E=\alpha -\pm 1\times \beta \nonumber \]
or
\[ E=\alpha \mp \beta \nonumber \]
Since \(\beta\) is negative, the two energies are ordered (Figure 10.5.4 )
- For \(\pi_1\): \(E_1 =\alpha + \beta\)
- For \(\pi_2\): \(E_2 =\alpha - \beta\)

To extract the coefficients attributed to these energies, the corresponding \(x\) values can be substituted back into the Secular Equations (Equation \(\ref{seceq}\)). For the lower energy state (\(x=-1\))
\[\begin{bmatrix} -1 & 1 \\ 1 & -1 \\ \end{bmatrix} \times \begin{bmatrix} c_1 \\ c_2 \\ \end{bmatrix}= 0 \nonumber \]
This gives \(c_1=c_2\) and the molecular orbitals attributed to this energy is then (based off of Equation \(\ref{LCAO}\)):
\[ |\psi_1 \rangle = N_1 (\phi_1 \rangle + | \phi_2 \rangle ) \label{HOMO} \]
where \(N_1\) is the normalization constant for this molecular orbital; this is the bonding molecular orbital.
For the higher energy molecular orbital (\(x=-1\)) and then
\[\begin{bmatrix} 1 & 1 \\ 1 & 1 \\ \end{bmatrix} \times \begin{bmatrix} c_1 \\ c_2 \\ \end{bmatrix}= 0 \nonumber \]
This gives \(c_1=-c_2\) and the molecular orbitals attributed to this energy is then (based off of Equation \(\ref{LCAO}\)):
\[ \psi_2 \rangle = N_2 (\phi_1 \rangle - | \phi_2 \rangle ) \label{LUMO} \]
where \(N_2\) is the normalization constant for this molecular orbital; this is the anti-bonding molecular orbital.
The normalization constants for both molecular orbitals can obtained via the standard normalization approach (i.e., \(\langle \psi_i | \psi_i \rangle =1\)) to obtain
\[N_1 = N_2 = \dfrac{1}{\sqrt{2}}\nonumber \]
These molecular orbitals form the \(\pi\)-bonding framework and since each carbon contributes one electron to this framework, only the lowest molecular orbital (\( | \psi_1 \rangle\)) is occupied (Figure 10.5.5 ) in the ground state. The corresponding electron configuration is then \( \pi_1^2\).

HOMO and LUMO are acronyms for highest occupied molecular orbital and lowest unoccupied molecular orbital, respectively and are often referred to as frontier orbitals. The energy difference between the HOMO and LUMO is termed the HOMO–LUMO gap.
The 3-D calculated \(\pi\) molecular orbitals are shown in Figure 10.5.6 .

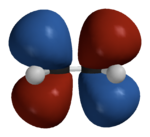
Hückel theory was developed in the 1930's when computers were unavailable and a simple mathematical approaches were very important for understanding experiment. Although the assumptions in Hückel theory are drastic they enabled the early calculations of molecular orbitals to be performed with mechanical calculators or by hand. Hückel Theory can be extended to address other types of atoms in conjugated molecules (e.g., nitrogen and oxygen). Moreover, it can be extended to also treat \(\sigma\) orbitals and this "Extended Hückel Theory" is still used today. Despite the utility of Hückel Theory, it is highly qualitative and we should remember the limitations of Hückel Theory:
- Hückel Theory is very approximate
- Hückel Theory cannot calculate energies accurately (electron-electron repulsion is not calculated)
- Hückel Theory typically overestimates predicted dipole moments
Hückel Theory is best used to provide simplified models for understanding chemistry and for a detailed understanding modern ab initio molecular methods discussed in Chapter 11 are needed.
Contributors
Seymour Blinder (Professor Emeritus of Chemistry and Physics at the University of Michigan, Ann Arbor)
- Wikipedia
- StackExchange (Philipp)