
2.2: Structure and Function- Proteins I
- Page ID
- 347409

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)Source: BiochemFFA_2_2.pdf. The entire textbook is available for free from the authors at http://biochem.science.oregonstate.edu/content/biochemistry-free-and-easy
Proteins are the workhorses of the cell. Virtually everything that goes on inside of cells happens as a result of the actions of proteins. Among other things, protein enzymes catalyze the vast majority of cellular reactions, mediate signaling, give structure both to cells and to multicellular organisms, and exert control over the expression of genes. Life, as we know it, would not exist if there were no proteins. The versatility of proteins arises because of their varied structures.
Proteins are made by linking together amino acids, with each protein having a characteristic and unique amino acid sequence. To get a sense for the diversity of proteins that can be made using 20 different amino acids, consider that the number of different combinations possible with 20 amino acids is 20n, where n=the number of amino acids in the chain. It becomes apparent that even a dipeptide made of just two amino acids joined together gives us 202 = 400 different combinations. If we do the calculation for a short peptide of 10 amino acids, we arrive at an enormous 10,240,000,000,000 combinations. Most proteins are much larger than this, making the possible number of proteins with unique amino acid sequences unimaginably huge.
Levels of Structure
The significance of the unique sequence, or order, of amino acids, known as the protein’s primary structure, is that it dictates the 3-D conformation the folded protein will have. This conformation, in turn, will determine the function of the protein. We shall examine protein structure at four distinct levels (Figure 2.17) - 1) how sequence of the amino acids in a protein (primary structure) gives identity and characteristics to a protein (Figure 2.18); 2) how local interactions between one part of the polypeptide backbone and another affect protein shape (secondary structure); 3) how the polypeptide chain of a protein can fold to allow amino acids to interact with each other that are not close in primary structure (tertiary structure); and 4) how different polypeptide chains interact with each other within a multi-subunit protein (quaternary structure).

At this point, we should provide a couple of definitions. We use the term polypeptide to refer to a single polymer of amino acids. It may or may not have folded into its final, functional form. The term protein is sometimes used interchangeably with polypeptide, as in “protein synthesis”. It is generally used, however, to refer to a folded, functional molecule that may have one or more subunits (made up of individual polypeptides). Thus, when we use the term protein, we are usually referring to a functional, folded polypeptide or peptides. Structure is essential for function. If you alter the structure, you alter the function - usually, but not always, this means you lose all function. For many proteins, it is not difficult to alter the structure.
Proteins are flexible, not rigidly fixed in structure. As we shall see, it is the flexibility of proteins that allows them to be amazing catalysts and allows them to adapt to, respond to, and pass on signals upon binding of other molecules or proteins. However, proteins are not infinitely flexible. There are constraints on the conformations that proteins can adopt and these constraints govern the conformations that proteins display.
Subtle changes
Even very tiny, subtle changes in protein structure can give rise to big changes in the behavior of proteins. Hemoglobin, for example, undergoes an incredibly small structural change upon binding of one oxygen molecule, and that simple change causes the remainder of the protein to gain a considerably greater affinity for oxygen that the protein didn’t have before the structural change.
Sequence, structure and function
As discussed earlier, the number of different amino acid sequences possible, even for short peptides, is very large. No two proteins with different amino acid sequences (primary structure) have identical overall structure. The unique amino acid sequence of a protein is reflected in its unique folded structure. This structure, in turn, determines the protein’s function. This is why mutations that alter amino acid sequence can affect the function of a protein.
Protein Synthesis
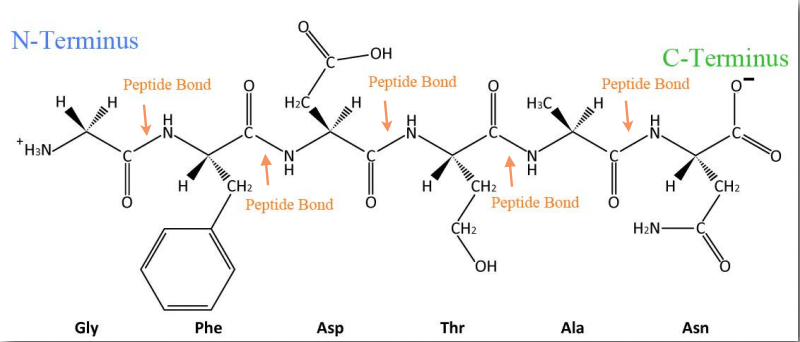
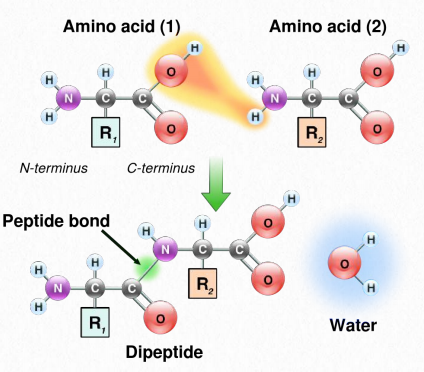
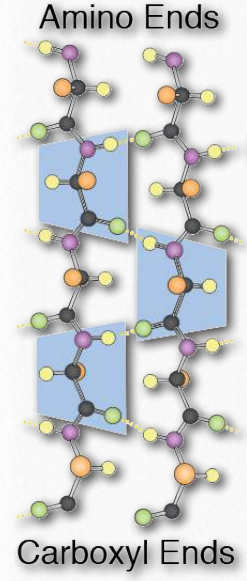
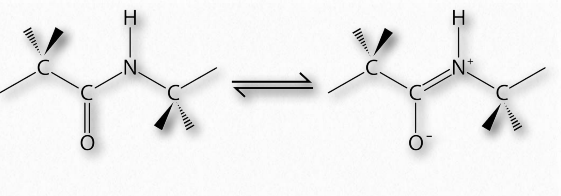
Synthesis of proteins occurs in the ribosomes and proceeds by joining the carboxyl terminus of the first amino acid to the amino terminus of the next one (Figure 2.19). The end of the protein that has the free α-amino group is referred to as the amino terminus or N-terminus. The other end is called the carboxyl terminus or C-terminus , since it contains the only free α-carboxyl group. All of the other α-amino groups and α-carboxyl groups are tied up in forming peptide Figure 2.19 Linking of amino acids through peptide bond formation bonds that join adjacent amino acids together. Proteins are synthesized starting with the amino terminus and ending at the carboxyl terminus.
Schematically, in Figure 2.18, we can see how sequential R-groups of a protein are arranged in an alternating orientation on either side of the polypeptide chain. Organization of R-groups in this fashion is not random. Steric hindrance can occur when consecutive R-groups are oriented on the same side of a peptide backbone (Figure 2.20)
Primary Structure
Primary structure is the ultimate determinant of the overall conformation of a protein. The primary structure of any protein arrived at its current state as a result of mutation and selection over evolutionary time. Primary structure of proteins is mandated by the sequence of DNA coding for it in the genome. Regions of DNA specifying proteins are known as coding regions (or genes).
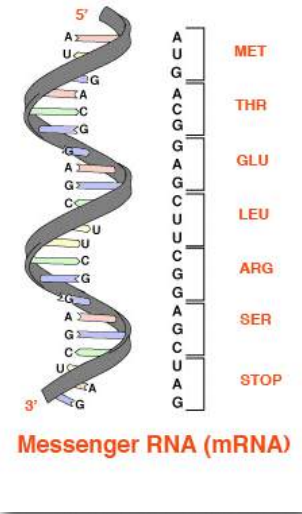
The base sequences of these regions directly specify the sequence of amino acids in proteins, with a one-to-one correspondence between the codons (groups of three consecutive bases) in the DNA and the amino acids in the encoded protein. The sequence of codons in DNA, copied into messenger RNA, specifies a sequence of amino acids in a protein. (Figure 2.21).
The order in which the amino acids are joined together in protein synthesis starts defining a set of interactions between amino acids even as the synthesis is occurring. That is, a polypeptide can fold even as it is being made. The order of the R-group structures and resulting interactions are very important because early interactions affect later interactions. This is because interactions start establishing structures - secondary and tertiary. If a helical structure (secondary structure), for example, starts to form, the possibilities for interaction of a particular amino acid Rgroup may be different than if the helix had not formed (Figure 2.22). R-group interactions can also cause bends in a polypeptide sequence (tertiary structure) and these bends can create (in some cases) opportunities for interactions that wouldn’t have been possible without the bend or prevent (in other cases) similar interaction possibilities.
Secondary Structure
As protein synthesis progresses, interactions between amino acids close to each other begin to occur, giving rise to local patterns called secondary structure. These secondary structures include the well known α- helix and β-strands. Both were predicted by Linus Pauling, Robert Corey, and Herman Branson in 1951. Each structure has unique features.
α-helix

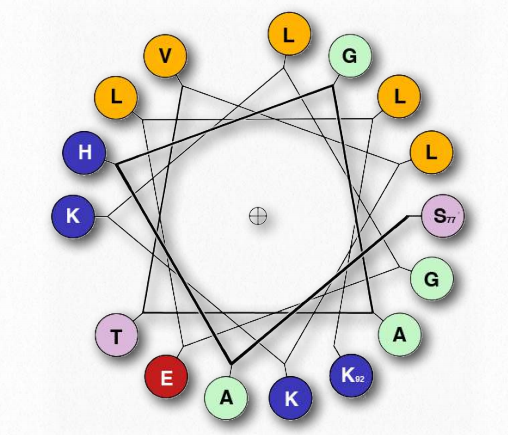
The α-helix has a coiled structure, with 3.6 amino acids per turn of the helix (5 helical turns = 18 amino acids). Helices are predominantly right handed - only in rare cases, such as in sequences with many glycines can left handed α- helices form. In the α-helix, hydrogen bonds form between C=O groups and N-H groups in the polypeptide backbone that are four amino acids distant. These hydrogen bonds are the primary forces stabilizing the α-helix.

We use the terms rise, repeat, and pitch to describe the parameters of any helix. The repeat is the number of residues in a helix before it begins to repeat itself. For an α-helix, the repeat is 3.6 amino acids per turn of the helix. The rise is the distance the helix elevates with addition of each residue. For an α-helix, this is 0.15 nm per amino acid. The pitch is the distance between complete turns of the helix. For an α-helix, this is 0.54 nm. The stability of an α-helix is enhanced by the presence of the amino acid aspartate.

β strand/sheet
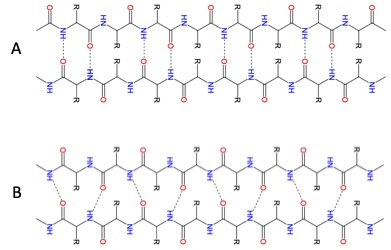
Figure 2.26 - β sheets: A. anti-parallel beta-sheet B parallel beta-sheet (from https://en.wikipedia.org/wiki/Beta_sheet)
In contrast to the compact coiled shape of the alpha-helix, beta-strands are more extended but contain bends that are sometimes referred to as pleats, like the pleats in a curtain. β-strands can be organized to form elaborately organized structures, such as sheets, barrels, and other arrangements.
β-sheets are stabilized by hydrogen bonds between carbonyl oxygen atoms and hydrogens of amine groups in the polypeptide backbone (Figure 2.28) from distant regions. In a higher order structure, strands can be arranged parallel (amino to carboxyl orientations the same) or anti-parallel (amino to carboxyl orientations opposite of each other (in Figure 2.27, the direction of the strand is shown by the arrowhead in the ribbon diagrams).
Turns
Turns (sometimes called reverse turns) are a type of secondary structure that, as the name suggests, causes a turn in the structure of a polypeptide chain. Turns give rise to tertiary structure ultimately, causing interruptions in the secondary structures (α- helices and β-strands) and often serve as connecting regions between two regions of secondary structure in a protein. Proline and glycine play common roles in turns, providing less flexibility (starting the turn) and greater flexibility (facilitating the turn), respectively.
There are at least five types of turns, with numerous variations of each giving rise to many different turns. The five types of turns are
• δ-turns - end amino acids are separated by one peptide bond
• γ-turns - separation by two peptide bonds
•β-turns - separation by three peptide bonds
•α-turns - separation by four peptide bonds
•π-turns - separation by five bonds
Of these, the β-turns are the most common form and the δ-turns are theoretical, but unlikely, due to steric limitations. Figure 2.29 depicts a β- turn.
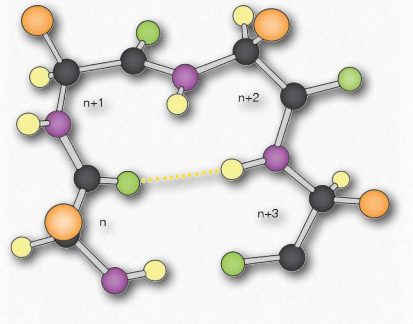
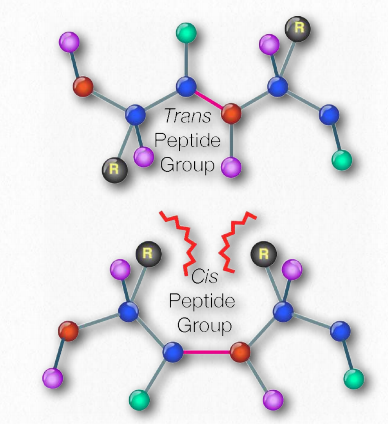
Notice that the arrangement of the backbone is limited by the partial double-bond character of the peptide bond: the backbone can exhibit free rotation around the bonds to the alpha-carbon of each amino acid residue, but not around the peptide bond itself.
Hydrophobicity
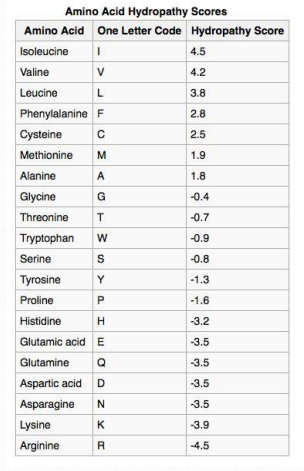
Table 2.4 - Hydropathy Scores
The chemistry of amino acid R-groups affects the structures they are most commonly found in. Subsets of their chemical properties can give clues to structure and, sometimes, cellular location. A prime example is the hydrophobicity (water-avoiding tendencies) of some R-groups. Given the aqueous environment of the cell, such R-groups are not likely to be on the outside surface of a folded protein.
However, this rule does not hold for regions of protein that may be embedded within the lipid bilayers of cellular/ organelle membranes. This is because the region of such proteins that form the transmembrane domains are buried in the hydrophobic environment in the middle of the lipid bilayer.
Not surprisingly, scanning primary sequences for specifically sized/spaced stretches of hydrophobic amino acids can help to identify proteins found in membranes. Table 2.4 shows hydrophobicity values for R-groups of the amino acids. In this set, the scale runs from positive values (hydrophobic) to negative values (hydrophilic).
[Note from AG: several sections of text were removed - please, ignore non-consecutive numbering of the figures here...the figures 2.31-2.36 are removed intentionally]
Random coils
Some sections of a protein assume no regular, discernible structure and are sometimes said to lack secondary structure, though they may have hydrogen bonds. Such segments are described as being in random coils and may have fluidity to their structure that results in them having multiple stable forms. Random coils are identifiable with spectroscopic methods and nuclear magnetic resonance (NMR) in which distinctive signals are observed.

Structural motifs = super-secondary structure
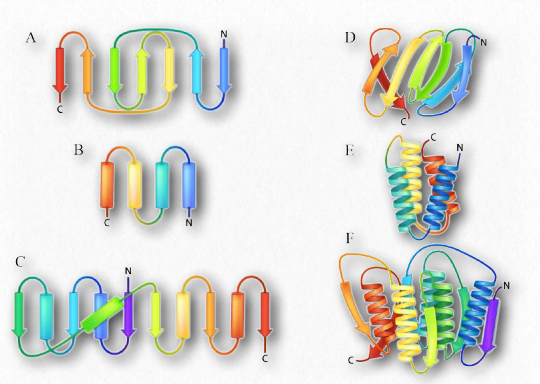
Another element of protein structure is harder to categorize because it incorporates elements of secondary and tertiary structure. Dubbed supersecondary structure (or structural motifs), these structures contain multiple nearby secondary structure components arranged in a specific way and that appear in multiple proteins. Since there are many ways of making secondary structures from different primary structures, so too can similar motifs arise from different primary sequences. An example of a structural motif is shown in Figure 2.37.
Tertiary structure
Proteins are distinguished from each other by the sequence of amino acids comprising them. The sequence of amino acids of a protein determines protein shape, since the chemical properties of each amino acid are forces that give rise to intermolecular interactions to begin to create secondary structures, such as α-helices and β-strands. The sequence also defines turns and random coils that play important roles in the process of protein folding.
Since shape is essential for protein function, the sequence of amino acids gives rise to all of the properties a protein has. As protein synthesis proceeds, individual components of secondary structure start to interact with each other, giving rise to folds that bring amino acids close together that are not near each other in primary structure (Figure 2.38). At the tertiary level of structure, interactions among the R-groups of the amino acids in the protein, as well as between the polypeptide backbone and amino acid side groups play a role in folding.
Globular proteins

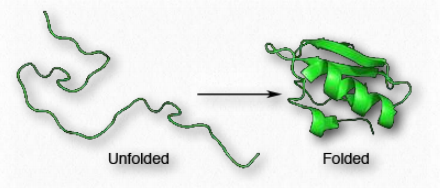
Folding gives rise to distinct 3-D shapes in proteins that are non-fibrous. These proteins are called globular. A globular protein is stabilized by the same forces that drive its formation. These include ionic interactions, hydrogen bonding, hydrophobic forces, ionic bonds, disulfide bonds and metallic bonds. Treatments such as heat, pH changes, detergents, urea and mercaptoethanol overpower the stabilizing forces and cause a protein to unfold, losing its structure and (usually) its function (Figure 2.39). The ability of heat and detergents to denature proteins is why we cook our food and wash our hands before eating - such treatments denature the proteins in the microorganisms on our hands. Organisms that live in environments of high temperature (over 50°C) have proteins with changes in stabilizing forces - additional hydrogen bonds, additional salt bridges (ionic interactions), and compactness may all play roles in keeping these proteins from unfolding.
Protein stabilizing forces
Before considering the folding process, let us consider some of the forces that help to stabilize proteins.
Hydrogen bonds
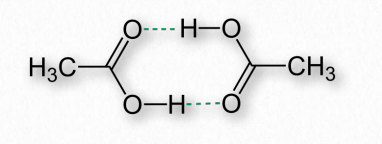
Hydrogen bonds arise as a result of partially charged hydrogens found in covalent bonds. This occurs when the atom the hydrogen is bonded to has a greater electronegativity than hydrogen itself does, resulting in hydrogen having a partial positive charge because it is not able to hold electrons close to itself (Figure 2.40).
Hydrogen partially charged in this way is attracted to atoms, such as oxygen and nitrogen that have partial negative charges, due to having greater electronegativities and thus holding electrons closer to themselves. The partially positively charged hydrogens are called donors, whereas the partially negative atoms they are attracted to are called acceptors. (See Figure 1.30).
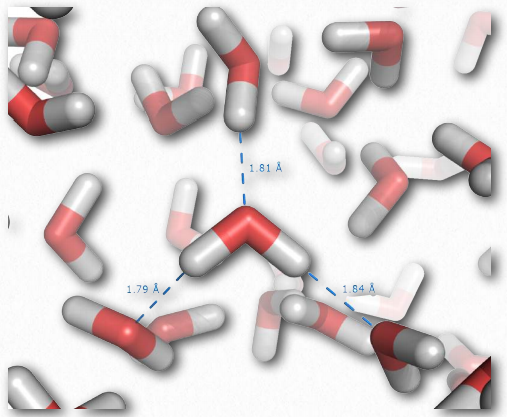
Individual hydrogen bonds are much weaker than a covalent bond, but collectively, they can exert strong forces. Consider liquid water, which contains enormous numbers of hydrogen bonds (Figure 2.41). These forces help water to remain liquid at room temperature. Other molecules lacking hydrogen bonds of equal or greater molecular weight than water, such as methane or carbon dioxide, are gases at the same temperature. Thus, the intermolecular interactions between water molecules help to “hold” water together and remain a liquid. Notably, only by raising the temperature of water to boiling are the forces of hydrogen bonding overcome, allowing water to become fully gaseous.
Hydrogen bonds are important forces in biopolymers that include DNA, proteins, and cellulose. All of these polymers lose their native structures upon boiling. Hydrogen bonds between amino acids that are close to each other in primary structure can give rise to regular repeating structures, such as helices or pleats, in proteins (secondary structure).
Ionic interactions
Ionic interactions are important forces stabilizing protein structure that arise from ionization of R-groups in the amino acids comprising a protein. These include the carboxyl amino acids (HERE), the amine amino acids as well as the sulfhydryl of cysteine and sometimes the hydroxyl of tyrosine.
Hydrophobic forces
Hydrophobic forces stabilize protein structure as a result of interactions that favor the exclusion of water. Non-polar amino acids (commonly found in the interior of proteins) favor associating with each other and this has the effect of excluding water. The excluded water has a higher entropy than water interacting with the hydrophobic side chains. This is because water aligns itself very regularly and in a distinct pattern when interacting with hydrophobic molecules.
When water is prevented from having these kinds of interactions, it is much more disordered that it would be if it could associate with the hydrophobic regions. It is partly for this reason that hydrophobic amino acids are found in protein interiors - so they can exclude water and increase entropy.
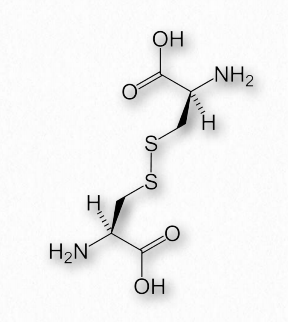
Disulfide bonds
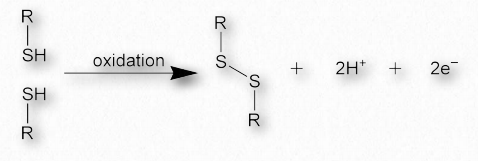
Disulfide bonds, which are made when two sulfhydryl side-chains of cysteine are brought into close proximity, covalently join together different protein regions and can give great strength to the overall structure (Figures 2.42 & 2.43). An Ode to Protein Structure by Kevin Ahern The twenty wee amino A's Define a protein many ways Their order in a peptide chain Determines forms that proteins gain And when they coil, it leaves me merry Cuz that makes structures secondary It's tertiary, I am told That happens when a protein folds But folded chains are downright scary When put together quaternary They're nature's wonders, that's for sure Creating problems, making cures A fool can fashion peptide poems But proteins come from ribosoems These joined residues of cysteine are sometimes referred to as cystine. Disulfide bonds are the strongest of the forces stabilizing protein structure.
van der Waals forces
van der Waals forces is a term used to describe various weak interactions, including those caused by attraction between a polar molecule and a transient dipole, or between two temporary dipoles. van der Waals forces are dynamic because of the fluctuating nature of the attraction, and are generally weak in comparison to covalent bonds, but can, over very short distances, be significant.
Post-translational modifications
Post-translational modifications can result in formation of covalent bonds stabilizing proteins as well. Hydroxylation of lysine and proline in strands of collagen can result in cross-linking of these groups and the resulting covalent bonds help to strengthen and stabilize the collagen.
Folding models
Two popular models of protein folding are currently under investigation. In the first (diffusion collision model), a nucleation event begins the process, followed by secondary structure formation. Collisions between the secondary structures (as in the β-hairpin in Figure 2.37) allow for folding to begin. By contrast, in the nucleation-condensation model, the secondary and tertiary structures form together.
Folding in proteins occurs fairly rapidly (0.1 to 1000 seconds) and can occur during synthesis - the amino terminus of a protein can start to fold before the carboxyl terminus is even made, though that is not always the case.
Folding process
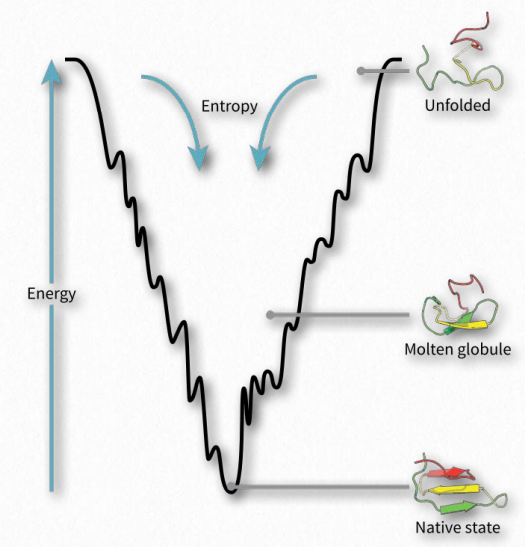
Protein folding is hypothesized to occur in a “folding funnel” energy landscape in which a folded protein’s native state corresponds to the minimal free energy possible in conditions of the medium (usually aqueous solvent) in which the protein is dissolved. As seen in the diagram (Figure 2.44), the energy funnel has numerous local minima (dips) in which a folding protein can become trapped as it moves down the energy plot. Other factors, such as temperature, electric/magnetic fields, and spacial considerations likely play roles.
If external forces affect local energy minima during folding, the process and end-product can be influenced. As the speed of a car going down a road will affect the safety of the journey, so too do energy considerations influence and guide the folding process, resulting in fully functional, properly folded proteins in some cases and misfolded “mistakes” in others.
Getting stuck
As the folding process proceeds towards an energy minimum (bottom of the funnel in Figure 2.44), a protein can get “stuck” in any of the local minima and not reach the final folded state. Though the folded state is, in general, more organized and therefore has reduced entropy than the unfolded state, there are two forces that overcome the entropy decrease and drive the process forward.
The first is the magnitude of the decrease in energy as shown in the graph. Since ΔG = ΔH -TΔS, a decrease in ΔH can overcome a negative ΔS to make ΔG negative and push the folding process forward. Favorable (decreased) energy conditions arise with formation of ionic bonds, hydrogen bonds, disulfide bonds, and metallic bonds during the folding process. In addition, the hydrophobic effect increases entropy by allowing hydrophobic amino acids in the interior of a folded protein to exclude water, thus countering the impact of the ordering of the protein structure by making the ΔS less negative.
Spectroscopy
Because of our inability to accurately predict tertiary structure based on amino acid sequence, proteins structures are actually determined using techniques of spectroscopy. In these approaches, proteins are subjected to varied forms of electromagnetic radiation and the ways they interact with the radiation allows researchers to determine atomic coordinates at Angstrom resolution from electron densities (by X-ray crystallography) and how nuclei spins interact (by NMR).
Diseases of protein misfolding
The proper folding of proteins is essential to their function. It follows then that misfolding of proteins (also called proteopathy) might have consequences. In some cases, this might simply result in an inactive protein. Protein misfolding also plays a role in numerous diseases, such as Mad Cow Disease, Alzheimers, Parkinson’s Disease, and CreutzfeldJakob disease. Many, but not all, misfolding diseases affect brain tissue.
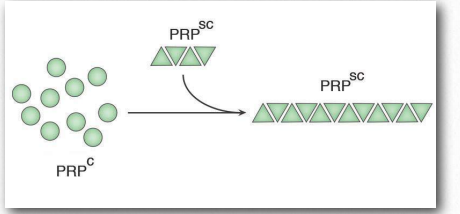
Insoluble deposits
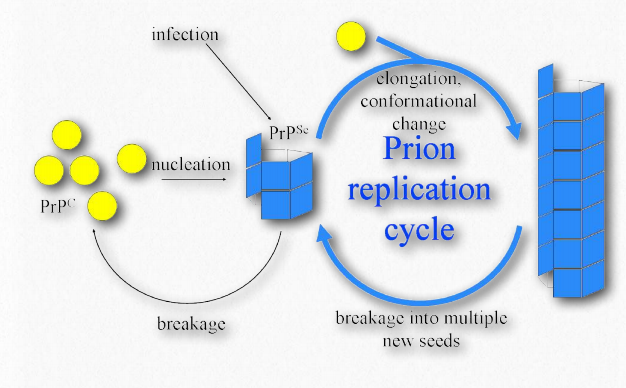
Misfolded proteins will commonly form aggregates called amyloids that are harmful to tissues containing them because they change from being soluble to insoluble in water and form deposits. The process by which misfolding (Figure 2.45) occurs is not completely clear, but in many cases, it has been demonstrated that a “seed” protein which is misfolded can induce the same misfolding in other copies of the same protein. These seed proteins are known as prions and they act as infectious agents, resulting in the spread of disease. The list of human diseases linked to protein misfolding is long and continues to grow. A Wikipedia link is HERE.
Prions
Prions are infectious protein particles that cause transmissible spongiform encephalopathies (TSEs), the best known of which is Mad Cow disease. Other manifestations include the disease, scrapie, in sheep, and human diseases, such as CreutzfeldtJakob disease (CJD), Fatal Familial Insomnia, and kuru. The protein involved in these diseases is a membrane protein called PrP. PrP is encoded in the genome of many organisms and is found in most cells of the body. PrPc is the name given to the structure of PrP that is normal and not associated with disease. PrPSc is the name given to a misfolded form of the same protein, that is associated with the development of disease symptoms (Figure 2.45).
Misfolded
The misfolded PrPSc is associated with the TSE diseases and acts as an infectious particle. A third form of PrP, called PrPres can be found in TSEs, but is not infectious. The ‘res’ of PrPres indicates it is protease resistant. It is worth noting that all three forms of PrP have the same amino acid sequence and differ from each other only in the ways in which the polypeptide chains are folded. The most dangerously misfolded form of PrP is PrPSc, because of its ability to act like an infectious agent - a seed protein that can induce misfolding of PrPc , thus converting it into PrPSc.
Function
The function of PrPc is unknown. Mice lacking the PrP gene do not have major abnormalities. They do appear to exhibit problems with long term memory, suggesting a function for PrPc . Stanley Prusiner, who discovered prions and coined the term, received the Nobel Prize in Medicine in 1997 for his work. I think that if I chanced to be on A protein making up a prion I’d twist it and for goodness sakes Stop it from making fold mistakes
Amyloids
Amyloids are a collection of improperly folded protein aggregates that are found in the human body. As a consequence of their misfolding, they are insoluble and contribute to some twenty human diseases including important neurological ones involving prions. Diseases include (affected protein in parentheses) - Alzheimer’s disease (Amyloid β), Parkinson’s disease (α-synuclein), Huntington’s disease (huntingtin), rheumatoid arthritis (serum amyloid A), fatal familial insomnia (PrPSc), and others.
Amino acid sequence plays a role in amyloidogenesis. Glutamine-rich polypeptides are common in yeast and human prions. Trinucleotide repeats are important in Huntington’s disease. Where sequence is not a factor, hydrophobic association between β-sheets can play a role.
Amyloid β

Amyloid β refers to collections of small proteins (36-43 amino acids) that appear to play a role in Alzheimer’s disease. (Tau protein is the other factor.) They are, in fact, the main components of amyloid plaques found in the brains of patients suffering from the disease and arise from proteolytic cleavage of a larger amyloid precursor glycoprotein called Amyloid Precursor Protein, an integral membrane protein of nerve cells whose function is not known. Two proteases, β-secretase and γ- secretase perform this function. Amyloid β proteins are improperly folded and appear to induce other proteins to misfold and thus precipitate and form the amyloid characteristic of the disease. The plaques are toxic to nerve cells and give rise to the dementia characteristic of the disease.
It is thought that aggregation of amyloid β proteins during misfolding leads to generation of reactive oxygen species and that this is the means by which neurons are damaged. It is not known what the actual function of amyloid β is. Autosomal dominant mutations in the protein lead to early onset of the disease, but this occurs in no more than 10% of the cases. Strategies for treating the disease include inhibition of the secretases that generate the peptide fragments from the amyloid precursor protein.
Huntingtin
Huntingtin is the central gene in Huntington’s disease. The protein made from it is glutamine rich, with 6-35 such residues in its wild-type form. In Huntington’s disease, this gene is mutated, increasing the number of glutamines in the mutant protein to between 36 and 250. The size of the protein varies with the number of glutamines in the mutant protein, but the wild-type protein has over 3100 amino acids and a molecular weight of about 350,000 Da. Its precise function is not known, but huntingtin is found in nerve cells, with the highest level in the brain. It is thought to possibly play roles in transport, signaling, and protection against apoptosis. Huntingtin is also required for early embryonic development. Within the cell, huntingtin is found localized primarily with microtubules and vesicles.
Trinucleotide repeat
The huntingtin gene contains many copies of the sequence CAG (called trinucleotide repeats), which code for the many glutamines in the protein. Huntington’s disease arises when extra copies of the CAG sequence are generated when the DNA of the gene is being copied. Expansion of repeated sequences can occur due to slipping of the polymerase relative to the DNA template during replication. As a result, multiple additional copies of the trinucleotide repeat may be made, resulting in proteins with variable numbers of glutamine residues. Up to 35 repeats can be tolerated without problem. The number of repeats can expand over the course of a person’s lifetime, however, by the same mechanism. Individuals with 36-40 repeats begin to show signs of the disease and if there are over 40, the disease will be present.
Molecular chaperones
The importance of the proper folding of proteins is highlighted by the diseases associated with misfolded proteins, so it is no surprise, then, that cells expend energy to facilitate the proper folding of proteins. Cells use two classes of proteins known as molecular chaperones, to facilitate such folding in cells. Molecular chaperones are of two kinds, the chaperones, and the chaperonins. An example of the first category is the Hsp70 class of proteins. Hsp stands for “heat shock protein”, based on the fact that these proteins were first observed in large amounts in cells that had been briefly subjected to high temperatures. Hsps function to assist cells in stresses arising from heat shock and exposure to oxidizing conditions or toxic heavy metals, such as cadmium and mercury. However, they also play an important role in normal conditions, where they assist in the proper folding of polypeptides by preventing aberrant interactions that could lead to misfolding or aggregation. The Hsp70 proteins are found in almost all cells and use ATP hydrolysis to stimulate structural changes in the shape of the chaperone to accommodate binding of substrate proteins. The binding domain of Hsp70s contains a β-barrel structure which wraps around the polypeptide chain of the substrate and has affinity for hydrophobic side chains of amino acids. As shown in Figure 2.50, Hsp70 binds to polypeptides as they emerge from ribosomes during protein synthesis. Binding of substrate stimulates ATP hydrolysis and this is facilitated by another heat shock protein known as Hsp40. The hydrolysis of ATP causes the Hsp70 to taken on a closed conformation that helps shield exposed hydrophobic residues and prevent aggregation or local misfolding.
After protein synthesis is complete, ADP is released and replaced by ATP and this results in release of the substrate protein, which then allows the full length polypeptide to fold correctly.

In heat shock
In times of heat shock or oxidative stress, Hsp70 proteins bind to unfolded hydrophobic regions of proteins to similarly prevent them from aggregating and allowing them to properly refold. When proteins are damaged, Hsp70 recruits enzymes that ubiquitinate the damaged protein to target them for destruction in proteasomes. Thus, the Hsp70 proteins play an important role in ensuring not only that proteins are properly folded, but that damaged or nonfunctional proteins are removed by degradation in the proteasome.
Chaperonins
A second class of proteins involved in assisting other proteins to fold properly are known as chaperonins. The best studied chaperonins are the GroEL/GroES complex proteins found in bacteria (Figure 2.51).
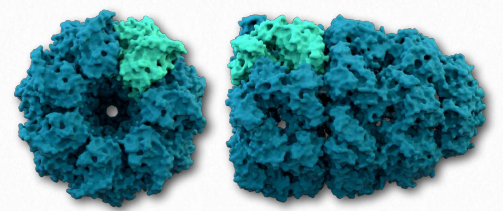
GroEL/GroES may not be able to undo aggregated proteins, but by facilitating proper folding, it provides competition for misfolding as a process and can reduce or eliminate problems arising from improper folding. GroEL is a double-ring 14mer with a hydrophobic region that can facilitate folding of substrates 15-60 kDa in size. GroES is a singlering heptamer that binds to GroEL in the presence of ATP and functions as a cover over GroEL. Hydrolysis of ATP by chaperonins induce large conformational changes that affect binding of substrate proteins and their folding. It is not known exactly how chaperonins fold proteins. Passive models postulate the chaperonin complex functioning inertly by preventing unfavorable intermolecular interactions or placing restrictions on spaces available for folding to occur. Active models propose that structural changes in the chaperonin complex induce structural changes in the substrate protein.
Protein breakdown
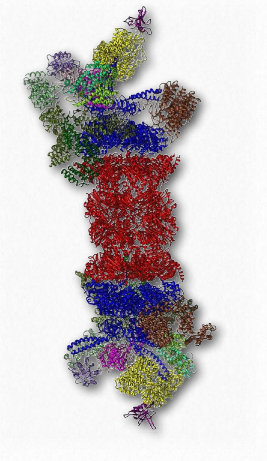
Another protein complex that has an important function in the lifetime dynamics of proteins is the proteasome (Figure 2.52). Proteasomes, which are found in all eukaryotes and archaeans, as well as some bacteria, function to break down unneeded or damaged proteins by proteolytic degradation. Proteasomes help to regulate the concentration of some proteins and degrade ones that are misfolded. The proteasomal degradation pathway plays an important role in cellular processes that include progression through the cell cycle, modulation of gene expression, and response to oxidative stresses.
Degradation in the proteasome yields short peptides seven to eight amino acids in length. Threonine proteases play important roles. Breakdown of these peptides yields individual amino acids, thus facilitating their recycling in cells. Proteins are targeted for degradation in eukaryotic proteasomes by attachment to multiple copies of a small protein called ubiquitin (8.5 kDa - 76 amino acids). The enzyme catalyzing the reaction is known as ubiquitin ligase. The resulting polyubiquitin chain is bound by the proteasome and degradation begins. Ubiquitin was named due to it ubiquitously being found in eukaryotic cells.
Ubiquitin
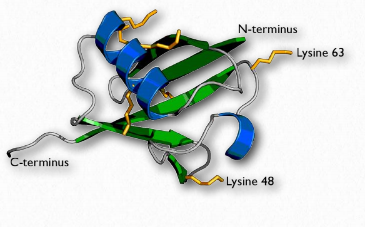
Ubiquitin (Figure 2.53) is a small (8.5 kDa) multi-functional protein found in eukaryotic cells. It is commonly added to target proteins by action of ubiquitin ligase enzymes (E3 in Figure 2.54). One (ubiquitination) or many (polyubiquitination) ubiquitin molecules may be added. Attachment of the ubiquitin is through the side chain of one of seven different lysine residues in ubiquitin.
The addition of ubiquitin to proteins has many effects, the best known of which is targeting the protein for degradation in the proteasome. Proteasomal targeting is seen when polyubiquitination occurs at lysines #29 and 48. Polyubiquitination or monoubiquitination at other lysines can result in altered cellular location and changed protein-protein interactions. The latter may alter affect inflammation, endocytic trafficking, translation and DNA repair.
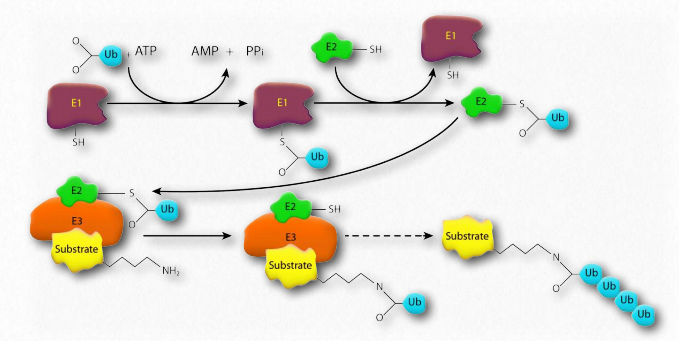
Ubiquitin ligase malfunction
Parkin is a Parkinson’s disease-related protein that, when mutated, is linked to an inherited form of the disease called autosomal recessive juvenile Parkinson’s disease. The function of the protein is not known, but it is a component of the E3 ubiquitin ligase system responsible for transferring ubiquitin from the E2 protein to a lysine side chain on the target protein. It is thought that mutations in parkin lead to proteasomal dysfunction and a consequent inability to break down proteins harmful to dopaminergic neurons. This results in the death or malfunction of these neurons, resulting in Parkinson’s disease.
Intrinsically disordered proteins
Movie 2.1 - Dynamic movement of cytochrome C in solution Wikipedia
As is evident from the many examples described elsewhere in the book, the 3-D structure of proteins is important for their function. But, increasingly, it is becoming evident that not all proteins fold into a stable structure. Studies on the so-called intrinsically disordered proteins (IDPs) in the past cou- ple of decades has shown that many proteins are biologically active, even thought they fail to fold into stable structures. Yet other proteins exhibit regions that remain unfolded (IDP regions) even as the rest of the polypeptide folds into a structured form.
Intrinsically disordered proteins and disordered regions within proteins have, in fact, been known for many years, but were regarded as an anomaly. It is only recently, with the realization that IDPs and IDP regions are widespread among eukaryotic proteins, that it has been recognized that the observed disorder is a "feature, not a bug".
Movie 2.2 SUMO-1, a protein with intrinsically disordered sections Wikipedia
Comparison of IDPs shows that they share sequence characteristics that appear to favor their disordered state. That is, just as some amino acid sequences may favor the folding of a polypeptide into a particular structure, the amino acid sequences of IDPs favor their remaining unfolded. IDP regions are seen to be low in hydrophobic residues and unusually rich in polar residues and proline. The presence of a large number of charged amino acids in the IDPs can inhibit folding through charge repulsion, while the lack of hydrophobic residues makes it difficult to form a stable hydrophobic core, and proline discourages the formation of helical structures. The observed differences between amino acid sequences in IDPs and structured proteins have been used to design algorithms to predict whether a given amino acid sequence will be disordered.
What is the significance of intrinsically disordered proteins or regions? The fact that this property is encoded in their amino acid sequences suggests that their disorder may be linked to their function. The flexible, mobile nature of some IDP regions may play a crucial role in their function, permitting a transition to a folded structure upon binding a protein partner or undergoing post-translational modification. Studies on several wellknown proteins with IDP regions suggest some answers. IDP regions may enhance the ability of proteins like the lac repressor to translocate along the DNA to search for specific binding sites. The flexibility of IDPs can also be an asset in protein-protein interactions, especially for proteins that are known to interact with many different protein partners.

For example, p53 has IDP regions that may allow the protein to interact with a variety of functional partners. Comparison of the known functions of proteins with predictions of disorder in these proteins suggests that IDPs and IDP regions may disproportionately function in signaling and regulation, while more structured proteins skew towards roles in catalysis and transport. Interestingly, many of the proteins found in both ribosomes and spliceosomes are predicted to have IDP regions that may play a part in correct assembly of these complexes. Even though IDPs have not been studied intensively for very long, what little is known of them suggests that they play an important and underestimated role in cells.
Metamorphic proteins
Another group of proteins that have recently changed our thinking about protein structure and function are the so-called metamorphic proteins. These proteins are capable of forming more than one stable, folded state starting with a single amino acid sequence. Although it is true that multiple folded conformations are not ruled out by the laws of physics and chemistry, metamorphic proteins are a relatively new discovery. It was known, of course, that prion proteins were capable of folding into alternative structures, but metamorphic proteins appear to be able to toggle back and forth between two stable structures. While in some cases, the metamorphic protein undergoes this switch in response to binding another molecule, some proteins that can accomplish this transition on their own. An interesting example is the signaling molecule, lymphotactin. Lymphotactin has two biological functions that are carried out by its two conformers- a monomeric form that binds the lymphotactin receptor and a dimeric form that binds heparin. It is possible that this sort of switching is more widespread than has been thought.
Refolding denatured proteins
All information for protein folding is contained in the amino acid sequence of the protein. It may seem curious then that most proteins do not fold into their proper, fully active form after they have been+++ denatured and the denaturant is removed. A few do, in fact. One good example is bovine ribonuclease (Figure 2.55). Its catalytic activity is very resistant to heat and urea and attempts to denature it don’t work very well. However, if one treats the enzyme with β-mercaptoethanol (which breaks disulfide bonds) prior to urea treatment and/or heating, activity is lost, indicating that the covalent disulfide bonds help stabilize the overall enzyme structure and when they are broken, denaturation can readily occur. When the mixture cools back down to room temperature, over time some enzyme activity reappears, indicating that ribonuclease re-folded under the new conditions.
Interestingly, renaturation will occur maximally if a tiny amount of β-mercaptoethanol is left in the solution during the process. The reason for this is because β- mercaptoethanol permits reduction (and breaking) of accidental, incorrect disulfide bonds during the folding process. Without it, these disulfide bonds will prevent proper folds from forming.
Irreversible denaturation
Most enzymes, however, do not behave like bovine ribonuclease. Once denatured, their activity cannot be recovered to any significant There are not very many ways Inactivating RNase It’s stable when it’s hot or cold Because disulfides tightly hold If you desire to make it stall Use hot mercaptoethanol extent. This may seem to contradict the idea of folding information being inherent to the sequence of amino acids in the protein. It does not.
Most enzymes don’t refold properly after denaturation for two reasons. First, normal folding may occur as proteins are being made. Interactions among amino acids early in the synthesis are not “confused” by interactions with amino acids later in the synthesis because those amino acids aren’t present as the process starts.
Chaperonins’ role
In other cases, the folding process of some proteins in the cell relied upon action of chaperonin proteins. In the absence of chaperonins, interactions that might result in misfolding occur, thus preventing proper folding. Thus, early folding and the assistance of chaperonins eliminate some potential “wrong-folding” interactions that can occur if the entire sequence was present when folding started.
Quaternary structure
A fourth level of protein structure is that of quaternary structure. It refers to structures that arise as a result of interactions between multiple polypeptides. The units can be identical multiple copies or can be different polypeptide chains. Adult hemoglobin is a good example of a protein with quaternary structure, being composed of two identical chains called α and two identical chains called β.
Though the α-chains are very similar to the β- chains, they are not identical. Both of the α- and the β-chains are also related to the single polypeptide chain in the related protein called myoglobin. Both myoglobin and hemoglobin have similarity in binding oxygen, but their behavior towards the molecule differ significantly. Notably, hemoglobin’s multiple subunits (with quaternary structure) compared to myoglobin’s single subunit (with no quaternary structure) give rise to these differences.
References
1. https://en.wikipedia.org/wiki/Van_der_W aals_force 105