
7.3: The phases of Drug Action
- Page ID
- 227599
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)The ability of a drug to carry its metabolic action (response) depends on two general phase. One phase is the ability of the drug to reach its site of action (receptor) in a particular cell. This process begins with the administration of the drug, its absorption, distribution, metabolization, and excretion through the body. This phase of drug action is called pharmacokinetics. Once at its action site, the ability of the drug to bind to the receptor depends on the chemical interactions between the chemical groups in the receptor and the drug (drug-receptor affinity). This phase of drug action is called pharmacodynamics. In order for a drug to be effective, it needs to exhibit acceptable pharmacokinetic and pharmacodynamic properties.

Pharmacokinetics and Pharmacodynamic stages of Drug Action. Image by Jorge Guerra Pires, CC BY-SA 4.0, via Wikimedia Commons
A. Pharmacokinetics
Pharmacokinetics deals with the absorption, distribution, biotransformation (metabolization), and excretion of drugs. These factors, coupled with dosage, determine the concentration of a drug at its sites of action and, hence, the intensity of its effects as a function of time. Many basic principles of biochemistry and enzymology and the physical and chemical principles that govern the active and passive transfer and the distribution of substances across biological membranes are readily applied to the understanding of this important aspect of medicinal chemistry.
Drug Absorption:
Absorption occurs after drugs enter the body and travel from the site of administration into the body’s circulation. Medications can enter the body through various routes of administration:
- oral (swallowing an aspirin tablet)
- enteral (administering to the GI tract such as via a NG tube)
- rectal (administering an acetaminophen [Tylenol] suppository)
- inhalation (breathing in medication from an inhaler)
- intramuscular (getting a flu shot in the deltoid muscle)
- subcutaneous (injecting insulin into the fat tissue beneath the skin)
- transdermal (wearing a nicotine patch)
Different factors, such as the chemical structure of the drug, the type of cellular tissue at the administration site, local pH, concentration (dosage), and the formulation of the drug (tablet, capsule, liquid, cream, etc) can affect the ability of the drug to enter the body.
Drug Distribution
Distribution is the process by which medication is distributed throughout the body. The distribution of a drug throughout the body is dependent on common factors such as blood flow, plasma protein binding, lipid solubility, the blood-brain barrier, and the placental barrier. Other factors include capillary permeability, differences between blood/tissue, and volume of distribution.
The bloodstream carries medications to their destinations in the body. Once the drug is in the bloodstream, a portion of it may exist as free drug, dissolved in plasma water. Some of the drug will be reversibly taken up by red cells, and some will be reversibly bound to plasma proteins. For many drugs, the bound forms can account for 95-98% of the total. This is important because it is the free drug that traverses cell membranes and produces the desired effect. It is also important because a protein-bound drug can act as a reservoir that releases the drug slowly and thus prolongs its action. With drug distribution, it is important to consider both the amount of free drug that is readily available to tissues, as well as the potential drug reserve that may be released over time.
Drug Metabolism
Once a drug has been absorbed and distributed in the body, it will then be broken down by a process known as metabolism. The breakdown of a drug molecule usually involves enzymes present in the liver. Many of the products of enzymatic breakdown, which are called metabolites, are less chemically active than the original molecule. Metabolites are usually more water-soluble than the parental drug and therefore easier to excrete in the urine.
Drug Excretion or Elimination
Drugs are eliminated from the body either unchanged or as metabolites. Most of the excretion is done by filtration at the kidneys, where a portion of the drug undergoes reabsorption back into the bloodstream, and the remainder is excreted in the urine. The liver also excretes byproducts and waste into the bile. Excretory organs eliminate polar compounds more efficiently than substances with high lipid solubility. Lipid-soluble drugs are thus not readily eliminated until they are metabolized to more polar compounds.
The kidney is the most important organ for elimination of drugs and their metabolites. Substances excreted in the feces are mainly unabsorbed orally ingested drugs or metabolites excreted in the bile and not reabsorbed from the intestinal tract. Excretion of drugs in milk is important not because of the amounts eliminated but because the excreted drugs are potential sources of unwanted pharmacological effects in the nursing infant. Pulmonary excretion is important mainly for the elimination of anesthetic gases and vapors: occasionally, small quantities of other drugs of metabolites are excreted by this route.
Drug elimination follows first-order kinetics. To illustrate first order kinetics we might consider what would happen if we were instantly inject (with an IV) a person with a drug, collect blood samples at various times and measure the plasma concentrations Cp of the drug. We might see a steady decrease in concentration as the drug is eliminated, as shown in the figure below.

Drug dosage and concentration
because of the pharmacokinetic phases, the plasma concentration (Cp) of a drug is not constant. As a drug is absorbed, metabolized, and eliminated, we see an onset, a peak and a duration of its effect. The plot below shows the effects of multiple doses given at different time intervals. Using a graph such as this, doctors can coordinate drug doses with proper time intervals in order to will keep drug concentrations at their optimum levels (between the blue and red lines, the therapeutic window)
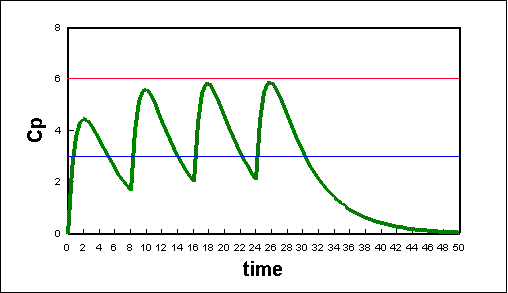
Plot of plasma concentration (Cp) due to multiple doses of a hypothetical drug.
Each dose was the same quantity of drug administrated every 8 hours
B. Pharmacodynamics: interaction of drugs with their sites of action
While there are several types of exceptions, the effects of most drugs result from their interaction with functional macromolecular components of the organism. Such interaction alters the function of the pertinent cellular component and thereby initiates the series of biochemical and physiological changes that are characteristic of the response to the drug. The term receptor is used to denote the component of the organism with which the chemical agent interacts. By virtue of interactions with such receptors, drugs do not create effects but merely modulate ongoing function. Thus, drugs cannot impart a new function to a cell.

Figure. - hypothetical drug in receptor site.
Structure-Activity
The affinity of a drug for a specific macromolecular component of the cell and its intrinsic activity are intimately related to its chemical structure. The relationship is frequently quite stringent, and relatively minor modifications in the drug molecule, particularly such subtle changes as stereochemistry, may result in major changes in pharmacological properties. Exploitation of structure-activity relationships has on many occasions led to the synthesis of valuable therapeutic agents. Since changes in molecular configuration need to alter all actions and effects of a drug equally, it is sometimes possible to develop a congener with a more favorable ratio of therapeutic to toxic effects, enhanced selectivity among different cells or tissues, or more acceptable secondary characteristics than those of the parent drug. In addition, effective therapeutic agents have been fashioned by developing structurally related competitive antagonists of other drugs or of endogenous substances known to be important in biochemical or physiological function. Minor modifications of structure can also have profound effects on the pharmacokinetic properties of drugs.
Key factors to consider in the structure of a drug are its polarity and molecular shape (stereochemistry).
Stereochemistry
Enantemerism can be produced by sp3 hybridized carbon atoms. Because free rotation about the chiral carbon is not possible, two stable forms of the molecule can exist. A molecule with two nonidentical asymmetric centers can exist as (22 = 4) four stereo isomers. Interaction with biological receptors can differ greatly between two enantomers, even to the point of no binding. There are numerous examples among drug molecules where only one isomer exhibits the desired pharmacology. Some isomers may even cause side effects or entirely different effects than its mirror image.
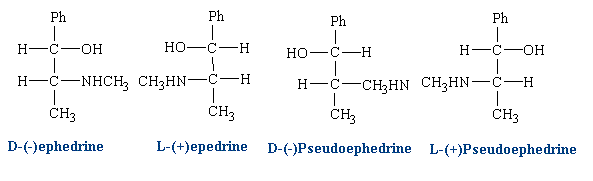
Ephedrine has two chiral centers and four isomers:
Different isomers can be used in different cases depending on the desired effect. Clinically, D(-) ephedrine is used to a large extent as an anti-asthmatic and, formerly, as a presser amine to restore low blood pressure as a result of trauma. L(+) pseudo-ephedrine is used primarily as a nasal decongestant.
| Isomer | Relative activity |
|---|---|
| D(-) Pseudoephedrine | 1 |
| L(+) Pseudoephedrine | 7 |
| L(+) Ephedrine | 11 |
| D(-) Ephedrine | 36 |
If the biological receptor has at least three binding sites, the receptor easily can differentiate enantomers (see figures below). The R(-)isomer has three points of interaction and is held in the conformation shown to maximize binding energy, whereas, the S(+)isomer can have only two sites of interaction.
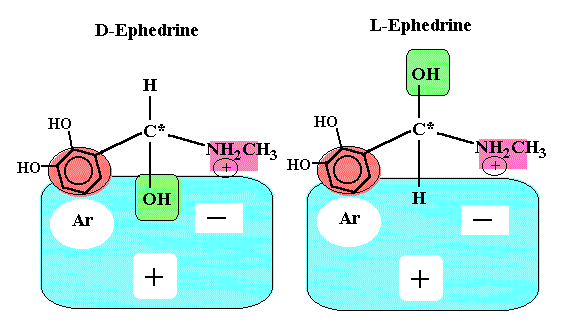
It should be noted that the structure of alpha and beta adrenergic receptors are not entirely known. Also we should not forget that there is also enantioselectivity with respect to pharmacokinetics, such as absorption, distribution, metabolism, and excretion.
Polarity
The affinity of a drug for its receptor is determined by the type and intensity of intermolecular forces between the functional groups present in the drug molecule and the amino acids in the receptors (most receptors are proteins). Hydrogen-bonding, dipole-dipole interactions, London Dispersion Forces, ion-dipole, etc., all contribute to increase the interactions between drug and receptor and increase the physiological response of the drug. At the same time, the presence of polar groups in the drug tend to increase water solubility and reduce the ability of a drug to permeate through the blood-brain barrier, while non-polar groups tend to increase lipid solubility and permeability to the blood-brain barrier
Sources
Ernstmeyer & Christman (Eds.) Chippewa Valley Technical College. Sourced from OpenRN. ChemLibreTexts content adapted under CC BY-NC-SA 3.0 license
Absorption. (2021, February 8). Retrieved April 6, 2021, from https://chem.libretexts.org/@go/page/24201
Distribution. (2021, February 8). Retrieved April 6, 2021, from https://chem.libretexts.org/@go/page/24202
Metabolism. (2021, February 8). Retrieved April 6, 2021, from https://chem.libretexts.org/@go/page/24203
Excretion. (2021, February 8). Retrieved April 6, 2021, from https://chem.libretexts.org/@go/page/24204
Gareth Thomas. Fundamentals of Medicinal Chemistry. Wiley-Blackwell; 2003.