
a) Linear Sweep Voltammetry
- Page ID
- 61486

In a previous section, we presented the diffusion controlled current-time behavior of a redox couple following a step to a potential sufficient to reduce all of the electroactive species near the surface of the electrode. Faradaic current was observed to decay as a function of t-1/2 according to the Cottrell equation. This was explained in terms of the concentration profile extending from the electrode surface into the bulk of solution. The thickness of this diffusion layer increases with time, decreasing the slope of the concentration gradient, and thus reducing the observed current.
We must now consider what happens to the concentration of electroactive species near the electrode as the potential is scanned, rather than stepped. Instead of going immediately to zero, the concentration of Ox is now dependent on the magnitude of the applied potential with respect to the E0’, with the ratio of CRed to COx (at 25 oC) given by the Nernst equation
\[\mathrm{E = E^0}\mathrm{’– (0.059 / n) \log [C_{Red}/ C_{Ox}]}\]
For our discussion, it is convenient to express the applied potential, E, in terms of its proximity to E0’, rearranging the Nernst equation as follows
\[\mathrm{\dfrac{n({E^0}'−E)}{0.059}=\log\dfrac{C_{Red}}{C_{Ox}}}\]
The concentration gradients existing for Ox and Red at several values of [n (E0' − E)] are illustrated in Figure 15 (0.059 V = 59 mV).
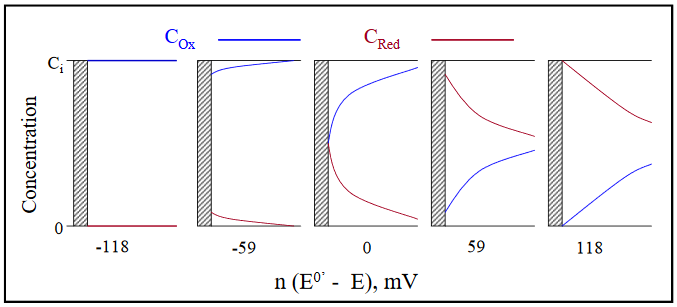
Figure 15
For a 1-electron reduction under diffusion control with an applied potential 118 mV positive of the E0’, COx = 100 CRed at the electrode surface. A potential 59 mV positive of the E0’ yields COx = 10 CRed. When the applied potential equals the E0’, (E0' − E) = 0, and the two concentrations are equal adjacent to the electrode, with each being half of the initial, or bulk concentration of COx (Ci in Figure 15). Beyond the E0’, the concentration of CRed becomes larger than that of COx, with CRed = 10 COx at a point 59 mV more negative than the E0’, and 100 COx at a point 118 mV more negative.
An important difference in the concentration gradients for potential sweep experiments when compared to large potential step experiments is that the slope of the gradient is dependent upon both the COx at the electrode surface (non-zero in the vicinity of E0’) and the thickness of the diffusion layer, not just the latter. A consequence of this is that the maximum concentration gradient (and hence maximum current) is observed at a potential 28.5 mV (for n = 1) beyond the E0’ of the redox couple. The potential – time and current – potential curves are presented in Figure 16 for a 1-electron, diffusion controlled redox couple with an E0’ of -0.300 V. The rate at which the potential is scanned is called the scan rate (big surprise) and is represented by v, generally with units of V/s. The initial potential is 0.00 V, with the final potential equal to -0.50 V. The current decrease beyond the peak potential (-0.328 V) is the result of COx at the electrode surface being driven to zero and the extension of the diffusion layer further and further into solution as the scan continues. (DigiElch simulation parameters: v = 0.100 V/s; A = 0.050 cm2; Ci = 1.0 mol/cm3; D = 1.0 x 10-5 cm2/s; α = 0.50; ks = 100 cm/s)
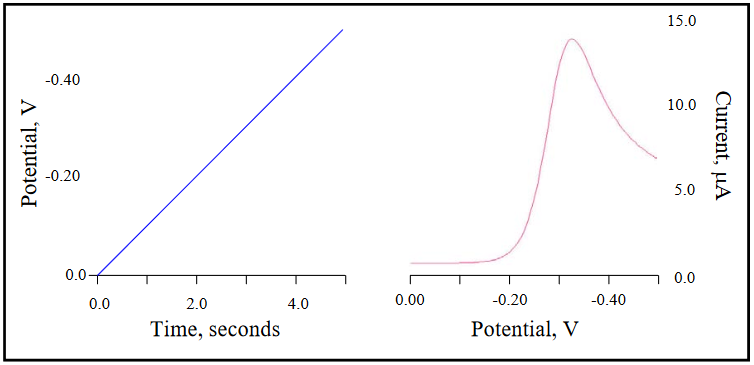
Figure 16
The observed peak current, ip, for a reversible electron transfer in LSV is given by the Randles-Sevcik equation
\[\mathrm{i_p = (2.69 \times 10^5)\, n^{3/2}\, A\, D^{1/2}\, C_i\,} v^{1/2}\]
where 2.69 x 10-5 is a collection of constants at 25 oC, and the variables are as previously defined. The capacitive current, which appears as the non-zero baseline at the left of the current-potential scan, must be subtracted from the total current to correctly obtain ip. Most commercial instrumentation allows for this to be done digitally.
LSV can be used to determine such parameters for a reversible system as E0’ , n, D, and Ci, while the use of a known redox couple can be employed in the calculation of A. As we have seen, the E0’ value occurs at a point (28.5/n) mV positive (for a reduction) of the observed peak potential, Ep. If difficulty is experienced with the accurate location of a peak potential, the potential measured at half the peak height (Ep/2) can be used to calculate it, lying (56.5/n) mV more positive than the Ep (for a reduction). If a value for D is known, the n-value for a redox active species can be calculated using the Randles-Sevcik equation. In most cases, even D values estimated from similar structures with known diffusion coefficients will suffice.
A plot of peak current, ip, as a function of v1/2, is linear for a redox active species under diffusion control, and can be a useful diagnostic indicator for a redox system being characterized for the first time. For reversible electron transfer, the position of Ep will not change with scan rate. The separation between E1/2 and Ep can also be used to diagnose reversible electron transfer, with |Ep – Ep/2| equal to (56.5/n) mV for a reversible electron transfer (ks > 0.02 cm/s). For irreversible electron transfer (ks < 5 x 10-5 cm/s), |Ep – Ep/2| will equal (47.7/αn) mV. Intermediate values for ks, defining the quasi-reversible regime, will yield intermediate values for |Ep – Ep/2| as well.