2.4: Cationic Polymerization
- Page ID
- 238853

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
Alkenes, or olefins, are probably the most common polymer feedstock. The pi bonds of alkenes are inherently nucleophilic. The addition of an electrophile to an alkene results in cleavage of the pi bond, with the electrons drawn to the electrophile, and consequently a cation can be left on one end of the former double bond.
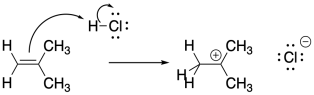
If the resulting cation interacts with another alkene, another electrophilic reaction occurs, and so on, leading to polymerization. Because of the presence of a key cationic intermediate along the reaction pathway, this method of polymerization is termed cationic polymerization. It is an example of a chain reaction that is initiated by a cationic species.
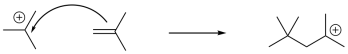
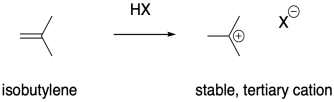
It stands to reason that alkenes that form stable cations would be particularly susceptible to polymerization via cationic methods. Cations are high-energy intermediates, so if a cation is relatively easy to form then the barrier for the process is lowered considerably. Isobutylene, (CH3)2C=CH2, is a good example of an alkene that would form a stable cation upon protonation. The relative stability of more-substituted cations dictates that the positive charge is found at a tertiary position.
Degree of substitution is just one way that carbocations can be stabilized. Conjugation is also a major factor in cation stability. Alkenes that give rise to delocalized cations are also good candidates for cationic polymerizaton. Styrene, PhCH=CH2, as well as butadiene, CH2=CHCH=CH2, and the related natural product isoprene, CH2=C(CH3)CH=CH2, can all be polymerized using cationic methods.
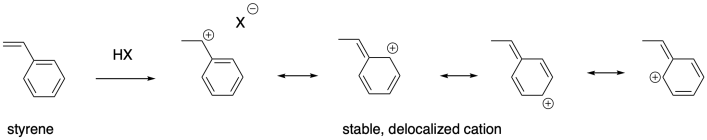
These examples are stabilized by allylic delocalization: the cation is part of a system that contains a C=C-C+ feature, which is delocalized as +C-C=C. Cations can also be stabilized via pi-donation. Oxygen and nitrogen are particularly good pi-donors towards carbocations, so oxygen and nitrogen substituents on alkenes render these compounds susceptible to cationic polymerization. Vinyl ethers, for example, can be polymerized this way.
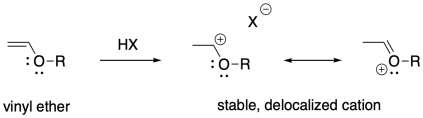
The obvious way to initiate these polymerizations is through addition of a strong acid. Protonation of the alkene should certainly result in formation of a carbocation. However, most common strong acids are mineral acids, typically found in aqueous media. The presence of nucleophilic water would quickly cap the carbocation before polymerization could take place.
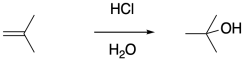
Of course, hydrogen chloride does not require water as a solvent. The addition of dry HCl gas to the olefin of interest should result in cation formation without subsequent hydration. Unfortunately, just the presence of the chloride ion, which can also act as a nucleophile, dooms polymerization to an early death.
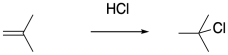
There are other, less nucleophilic acids that are not as strongly prone to classical addition across an alkene compared to the extent that we associate with HCl. Sulfuric acid and various sulfonic acids come to mind. Even in these cases, however, polymerization is limited by eventual collapse of the nucleophile with the cationic chain end.
Instead of using strong Brønsted acids to inititate polymerization, it is actually very common to use Lewis acids. Lewis acids cannot initiate cationic polymerization directly, but they can co-initiate the reaction in the presence of a water impurity, for example. The Lewis acid activates the water towards release of a proton. The proton then initiates the reaction. Traces of HCl can also act as the initiator when a Lewis acid co-initiator is employed, because the Lewis acid ties up the chloride ion so that it cannot connect with the growing cationic chain end.
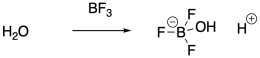
Protic initiators are not strictly required for cationic polymerization. Strong Lewis acids are also capable of activating alkyl halides. The carbocation that results can then initiate the polymerization chain reaction.
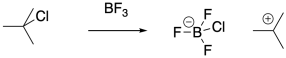
Even in these systems, there are chain-termination events that destroy the reactive intermediate and stop polymerization. As indicated previously, combination of a nucleophilic anion with the cationic chain end is a common fate of growing polymers. Halide ions, released in equilibrium from the Lewis acid-base complex, are still a possible problem, even though this event is much less likely in the presence of Lewis acidic co-initiators. Other, similar anions, such as hydroxide anions in water-initiated polymerizations, can play a similar role.

Anionic species can also carry out beta-eliminations on cationic chain ends. Beta-elimination results in the loss of the cationic intermediate, replacing it with a cain-end elkene. That event results in the production of a new initiator, and although it could be argued that the initiator might just re-initiate the same polymer chain, given relative concentrations it is much more likely that the initiator transfers the proton to a new monomer. In that case, a new growing chain starts from scratch and begins to accumulate monomers.
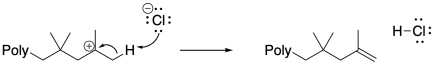
Surprisingly, chain-transfer events are another common mode of termination. In these cases, a hydride must be transferred from one chain to another, or from one position along the backbone to the growing chain end. This unusual hydride transfer is probably facilitated by very close contact distance between two chains, or between two segments of one chain, in a concentrated polymerization milieu. As a result, one chain end stops growing, and the chain begins growing from a new position along its backbone. This event results in chain branching, changing the morphology and properties of the polymer.
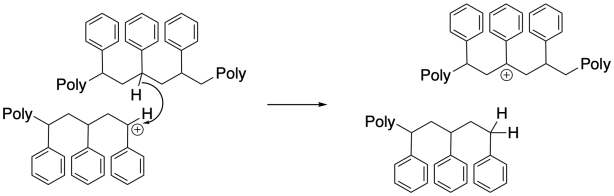
Normally, assuming a relatively rapid initiation step (formation of a cation from the first monomer), growth of tandem polymer chains will proceed at roughly the same rate, so that all of the chains reach similar lengths by the time all monomers have been consumed. What happens if some of those chains die prematurely? Other chains continue to grow to longer lengths. As a result, there is a wider distribution of chain lengths, or molecular weights, than there would be otherwise. A wider molecular weight distribution means that physical properties of the material may be less reproducible.
In a typical polymerization process, the reaction is followed by quenching, in which some agent is added that will stop further polymerization. In cationic polymerizations, quenching might be accomplished through addition of aqueous acid, because either the water or the conjugate of the acid will destroy the cation. The material is usually precipitated in a solvent that can dissolve any remaining monomer, but not polymer. Because some chains may have died very early, they might also remain in solution, leaving only larger chains in the precipitate.
Exercise 2.4.1
Choose the most stable cation from each of the following groups. Provide a reason for the stability of that cation.
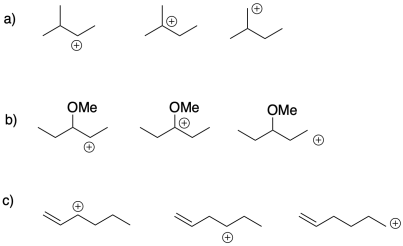
Exercise 2.4.2
In each group, select the alkene most suitable for cationic polymerization.
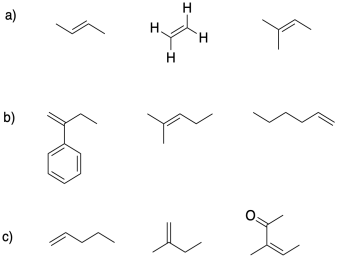
Exercise 2.4.3
Provide a mechanism for the formation of a protic initiator from the interaction of boron trifluoride with water.
Exercise 2.4.4
Anethole in a naturally-occuring compound that has been used in cationic polymerizations. Show why anethole should be a good monomer for this method.
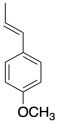
Exercise 2.4.5
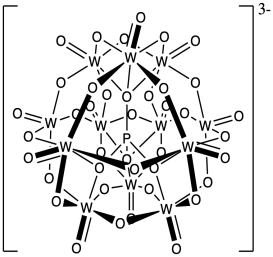
Phosphotungstates such as H3PW12O40 have been used as cationic initiators and do not show much termination by the counterion, below. Propose a reason for the low nucleophilicity of this counterion for the growing cationic chain.