
2.2: Pd-Catalyzed Cross Coupling Reactions
- Page ID
- 366590
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)After completing this section, you should be able to:
- Identify common Pd-catalyzed carbon-carbon bond forming reactions
- Draw and understand reaction mechanisms
- Use reactions in synthesis problems
Key Terms
Make certain that you can define, and use in context, the key terms below.
- Stille reaction
- Suzuki reaction
- Sonogashira reaction
- Heck reaction
- Tsuji-Trost reaction
- Buchwald-Hartwig amination
Study Notes
Palladium catalyzed reactions are the most common transition metal catalyzed cross coupling reactions. Their importance was recognized with the 2010 Nobel Prize for Richard Heck, Ei-ichi Negishi, and Akira Suzuki. These reactions provide straightforward methods for the construction of Csp2-Csp2, Csp2-Csp, and Csp2-N bonds. We will see many examples of these types of bond formations, and several others, throughout this chapter. While learning this material, don't forget to contrast Pd catalyzed reactions with popular intro orgo carbon-carbon bond forming reactions like Grignard or organolithium reactions with carbonyls and epoxides. At least one component of these reactions was sp3 hybridized, so it was impossible to generate the types of bonds that are formed most commonly by Pd catalyzed reactions. Adding Pd catalyzed reactions to your organic chemistry toolbox will greatly expand the types of molecules that you are able to synthesize.
Content
This chapter will focus on learning and applying common Pd catalyzed reactions for the formation of 1) Carbon-Carbon bonds: Stille, Suzuki, Sonogashira, and Heck 2) Carbon-Carbon or Carbon-Oxygen bonds: Tsuji-Trost and 3) Carbon-Nitrogen bonds: Buchwald-Hartwig. If you haven't already, please refer to the previous chapter for a discussion of transition metal structure and bonding along with common mechanistic steps in transition metal catalyzed reactions.
For all of the reactions in this chapter, we will depict the palladium catalyst as LnPd(0) to acknowledge that some number of ligands are bound to Pd but their actual number and structure are unimportant to the mechanism. Two things happen when a Pd complex dissolves in the reaction solvent. First, bonds to phosphine ligands break to establish an equilibrium with complexes having fewer ligands and open coordination sites for reactions to occur. Second, some reactions will begin with Pd(II) reagents. For example, most Sonogashira reactions are run with PdCl2(PPh3)2. These Pd reagents are reduced under the reaction conditions by several possible mechanisms that we won't concern ourselves with here. The net result is that Pd(0) is produced in the flask, and this complex can then catalyze the desired reactions. Even though we will generally ignore the phosphine ligands, the structure of these molecules is critically important to the success of Pd catalyzed reactions. Many of the recent advances in this field were made possible by the development of novel phosphine ligands. The Buchwald-Hartwig reaction is one such example.
Stille Reaction
The Stille reaction is the simplest Pd catalyzed cross coupling reaction where an organohalide combines with an organotin in the presence of a Pd(0) catalyst to form a new C-C bond. To avoid side reactions and production of mixtures, both components generally have Csp2 or Csp atoms bonded to the halogen and the tin. An example reaction and mechanism are shown below. Oxidative addition of bromobenzene with the Pd catalyst yields the first Pd(II) intermediate. Transmetallation transfers the alkene from Sn to Pd, generating the tributyltin bromide byproduct and the desired Pd(II) intermediate with two C-Pd bonds. Finally, reductive elimination generates the desired product and regenerates the Pd(0) catalyst.

A useful variant of the standard Stille reaction is the carbonylative Stille reaction where a carbonyl can be added between the organohalide and organotin components. This is made possible by running the reaction under an atmosphere of CO gas. An example with the carbonylative mechanism is shown below. After oxidative addition, CO associates with Pd forming a C-Pd bond. Next, the phenyl ligand can perform a carbonyl insertion step (similar to the alkene migratory insertion shown in the previous chapter) to form one new C-C bond. We now return to the regular Stille mechanism with the transmetallation and reductive elimination steps producing the final product and regenerating the catalyst.

Complete the following synthesis in 2 steps, one of which is a Stille reaction.

- Answer
-
Thinking retrosynthetically, we must be forming the single bond between the alkenes using a Stille reaction. That means we need to convert the ketone into a vinyl halide or halide equivalent in the first step. This is easily done by forming an enolate and adding triflic anhydride to make a vinyl triflate. As explained in the previous chapter, vinyl triflates participate readily in transition metal catalyzed reactions. So, our two step synthesis is 1) Deprotonation with LDA (to ensure we form the least substituted kinetic enolate) followed by addition of triflic anhydride to yield the vinyl triflate. 2) Stille reaction with vinyl tributyltin to generate the target.

Using retrosynthetic analysis, how would you make this target in one step using a Stille reaction?

- Answer
-
The key bond is the one between the alkene and the alkyne. This is the bond we will make in the forward direction using the Stille reaction.

Propose a 1 step synthesis of the target using a Stille reaction.

- Answer
-
This is the perfect molecule to use the carbonylative Stille reaction. We can start with a vinyl bromide and an alkynyl tin. Combining them with the palladium catalyst under a CO atmosphere yields the target.

Suzuki Reaction
The Suzuki reaction is similar to the Stille reaction but with boron used instead of tin for the transmetallation step. It also differs from the Stille reaction because Suzuki reactions require the presence of a base to activate the boron reagent prior to transmetallation. As shown below, the mechanistic steps are the same for these two popular transformations with the addition of the base promoted boron activation step that forms the reactive borate (negatively charged boron) compound. This borate reagent can be formed by a variety of oxygen bases in addition to carbonate like hydroxide and alkoxides (e.g., ethoxide, t-butoxide).

There are several advantages of the Suzuki reaction. First, we are already familiar with forming C-B bonds from intro orgo using a hydroboration reaction. Previously, we always followed this step with an oxidation reaction to form an alcohol. Now we can form the organoboron compound and use it in a Suzuki reaction. We can react alkenes or alkynes in hydroboration reactions to yield organoboron reagents for transmetallation. Several examples are shown below. Hydroboration of terminal alkynes is synthetically useful (internal alkynes yield product mixtures) when using hindered boranes like disiamylborane or catecholborane. Like all hydroborations, the reactions are syn additions with the larger boron adding to the less hindered side of the alkyne resulting in trans alkene products. To generate alkylboron reagents for use in Suzuki reactions, the most common reagent is 9-BBN (pictured below). A second advantage is the Suzuki reaction provides a way to use Pd catalysis to easily make Csp2-Csp3 or Csp-Csp3 bonds which can be challenging to make with other Pd catalyzed reactions. Hydroboration of an alkene provides the requisite B-Csp3 reagent (the 9-BBN derivative shown below) that participates in transmetallation then reductive elimination to yield the desired bond. We will see an example of this in one of the problems below.

What is the product of the follow reaction sequence?

- Answer
-
The first step is a hydroboration reaction with 9-BBN which is selective for the much less hindered terminal alkene. This sets up an example of a very synthetically useful intramolecular Suzuki reaction to form a 6-membered ring.

Propose a synthesis of the following target starting with compounds containing 6 carbons or fewer. One of your starting materials must be an alkyne.

- Answer
-
Thinking retrosynthetically, we can split the molecule between the two alkenes into two 6-carbon fragments that can be combined using a Suzuki reaction. The boron-containing component can then be simplified to the requisite alkyne starting material. In the forward direction, we start with 1-hexyne and hydroborate it with catecholborane (a 6-carbon reagent) to yield the vinylborane that participates in a Suzuki reaction with our 5-membered ring vinyl bromide to yield the target.

Sonogashira Reaction
The Sonogashira reaction enables the combination of an unsaturated carbon-containing halide or triflate with a terminal alkyne to yield a new Csp-Csp or Csp-Csp2 bond. One interesting aspect of this reaction is that it is catalyzed by a combination of Pd (to form the C-C bond) and Cu (to activate the terminal alkyne for transmetallation). Another key reagent is an amine base that promotes formation of the copper acetylide that participates in the transmetallation step. An example Sonogashira reaction and the mechanism is shown below. This is a very useful reaction and an important addition to our synthesis toolbox.

The previous three reactions, Stille, Suzuki, and Sonogashira, share a similar mechanism that includes oxidative addition, transmetallation, and reductive elimination steps. Other reactions with different metals have a similar catalytic cycle with palladium. Although we won't discuss them here, you are well equipped to understand these reactions when you see them in the literature. Two of the most common examples are the Negishi reaction (transmetallation with zinc) and the Kumada reaction (transmetallation with magnesium).
Heck Reaction
The Heck reaction proceeds via a different mechanistic pathway than the Stille, Suzuki, or Sonogashira reactions because it does not contain transmetallation or reductive elimination steps. Instead, the Heck reaction relies on an alkene insertion step for carbon-carbon bond formation and beta hydrogen elimination to generate the product. (As a reminder, these steps were described in detail in the previous chapter.) An example of the reaction and its mechanism are shown below. A few key points about the mechanism are worth highlighting. The alkene insertion step usually places the Pd at the more substituted position and the carbon at the least substituted position. This step is a syn addition to the alkene which necessitates a bond rotation in the next step to place a beta hydrogen syn to the Pd. There are two possible Hs that can end up syn but the conformation that places the phenyl and the ketone anti is preferred. (Note that for this mechanism, only one beta carbon has Hs. The other beta carbon is the ketone. In many reactions, there are several alkenes that can form. Heck reactions generally produce the most stable possible alkene product.) This conformation yields the more stable trans alkene product in the beta hydrogen elimination step. The Pd(0) catalyst is regenerated in the final deprotonation step that explains the need for base in the mechanism.

Heck reactions are commonly run intermolecularly, between two different reactants. However, the true power of the Heck reaction for synthesis becomes apparent when studying intramolecular Heck reactions. This is a very useful technique for quickly building molecular complexity. We will see an example of this in one of the problems below.
Predict the product of the following intermolecular Heck reaction.

- Answer
-
This is a tricky problem. Most students quickly predict the conjugated diene shown below that doesn't form. This is a good illustration of the importance of evaluating potential products formed from the beta hydrogen elimination step. In this reaction, there are two beta carbons with Hs that can be eliminated with Pd. One option, the beta H on the left, yields the conjugated diene. The other option, the beta H on the right, yields an enol product that tautomerizes to the observed aldehyde product. Carbonyls are more stable than alkenes, so this thermodynamic difference drives the reaction to the aldehyde.
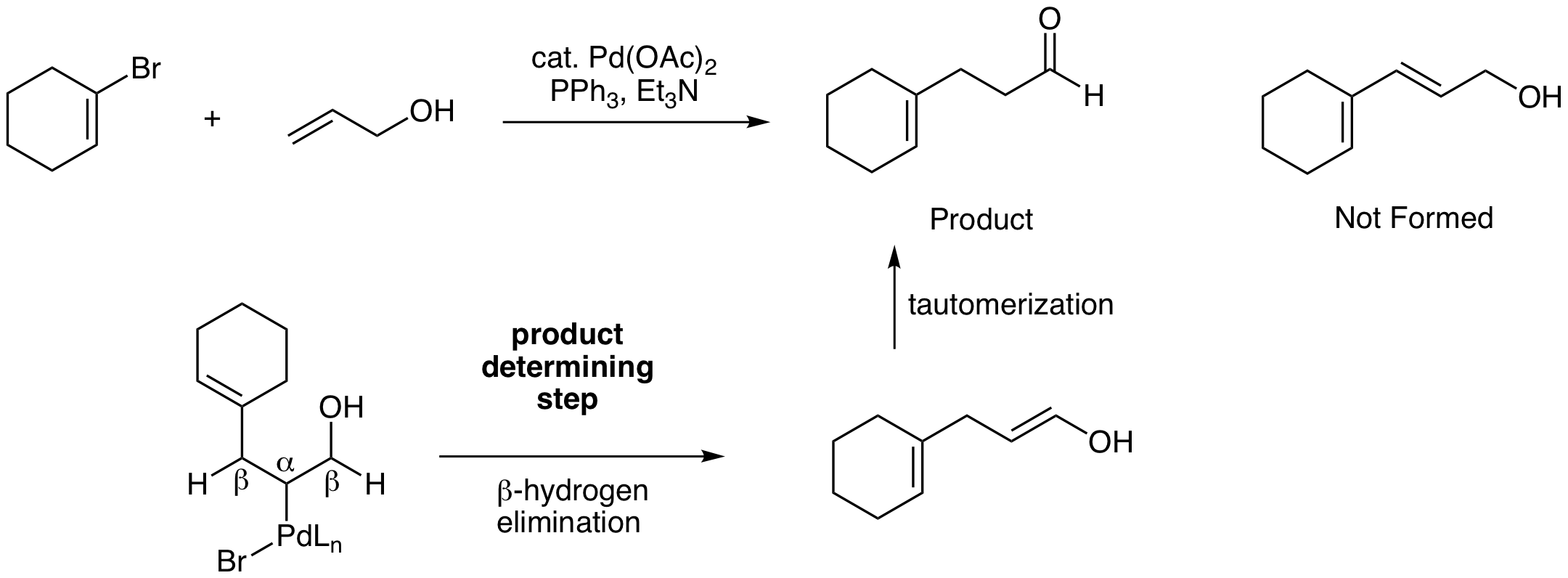
Predict the product of the following intramolecular Heck reaction.

- Answer
-
This is an excellent example of what is known as a Heck zipper reaction where multiple alkene insertions occur to form more than one ring. Working through the mechanism, we can see what is happening. After the initial oxidative addition step, the first alkene insertion occurs to form the new 6-membered ring. Normally, we would next perform a beta hydrogen elimination reaction to generate the product. However, this molecule has a quaternary carbon at the beta position, so beta hydrogen elimination is impossible. When that happens, the molecule undergoes another alkene insertion to form a second new ring. In this case, a new five membered ring. We again look to find a cis beta hydrogen. That is now possible when the Pd is on carbon #11 and the final product is formed.

Tsuji-Trost Reaction
The Tsuji-Trost reaction is a highly useful transformation that enables formation of C-O, C-N, and C-C bonds. The key reaction components are an alkene with an allylic leaving group (halide, acetate, or epoxide) and a nucleophile which is an alcohol, an amine, or a carbon with an acidic proton (think 1,3-dicarbonyl). An example with a mechanism is shown below. Reaction of allyl acetate with the 1,3-diketone nucleophile and a palladium catalyst yields the product with a new C-C bond to the allyl group. In the mechanism, which is unlike Pd catalyzed reactions we have already seen, the first step is alkene association. Next, Pd substitutes for the leaving group, generating a pi-allyl Pd complex. The acetate leaving group is now free to act as a base, deprotonating the acidic proton on the nucleophile. The resulting stabilized enolate adds to one of the terminal carbons on the pi-allyl complex, forming the new C-C bond. The final step is alkene dissociation that yields the product and regenerates the catalyst.

There are several important details to consider for the Tsuji-Trost reaction. First, it can happen inter- or intramolecularly, and we will see examples of each in the problems below. We also must consider regiochemistry and stereochemistry, depending on the structure of the reactants. One example below demonstrates that regioselectivity is governed by sterics, with the nucleophile adding to the less substituted side of the pi-allyl Pd complex. The other example below shows that the reaction is stereospecific, with substitution occurring with retention. As we saw previously in intro orgo, the only way for this to happen is for the mechanism to include two SN2 reactions. (Remember, SN1 reactions always yield racemization, equal amounts of retention and inversion.) The two SN2 reactions are when the Pd displaces the leaving group and when the nucleophile adds to the pi-allyl complex.

Predict the product of this intermolecular Tsuji-Trost reaction.

- Answer
-
The pi-allyl Pd complex is formed by breaking open the lactone. Next, the deprotonated diester adds to the least hindered side to yield the product.

Predict the product of this intramolecular Tsuji-Trost reaction.

- Answer
-
The Pd adds from the back to yield the pi-allyl complex. The enolate nucleophile adds from the front to form the new 6-membered ring.

Predict the product of this reaction and provide a mechanism to explain its formation.

- Answer
-
This is an example of an allylic epoxide participating in the Tsuji-Trost reaction. The Pd pi-allyl complex forms by inversion. A deprotonation step generates the carbanion nucleophile that adds with inversion to the least hindered side of the pi-allyl complex.

Buchwald-Hartwig Amination
The Buchwald-Hartwig amination was developed by Steve Buchwald (MIT) and John Hartwig (Berkeley) in the late 1990s. It has evolved into one of the most popular methods for the construction of aryl amines which are common functional groups in pharmaceutical compounds and natural products. The reaction combines an aryl halide, a primary or secondary amine, and a base in the presence of a palladium catalyst. As shown in the example below, specialized phosphine ligands pioneered by the Buchwald lab are often critical to the success of these reactions. The mechanism is similar to what we saw for the Stille, Suzuki, and Sonogashira reactions in that it starts with an oxidative addition and ends with a reductive elimination. The middle steps are different and involve the amine associating with Pd before the base deprotonates the amine proton resulting in the loss of halogen from the Pd.

Propose a product for this intramolecular Buchwald-Hartwig amination.

- Answer
-
This is an excellent strategy to form a new heterocyclic ring. In this case, it's a five membered ring that is part of the bicyclic dihydroindole system.

Summary Problems
Use what we have learned about Pd catalyzed reactions to solve these summary problems. You will also need to incorporate reactions from intro orgo and the pericyclic reactions chapter to complete some of the problems.
Complete the following synthesis using the indicated starting materials and any other compounds you would like. Hint: Start by finding the retron in the target and going backwards at least one step.

- Answer
-
The key is to start by identifying the cyclohexene Diels-Alder retron in the target. This simplifies the molecule and reveals a Stille retron, the single bond between the alkene and benzene ring. The final disconnection is the amide bond formation. In the forward direction, the carboxylic acid is activated as an acid chloride before adding the starting primary amine and pyridine. The next step is a Pd catalyzed Stille reaction between the aryl iodide and vinyl tin compound. The resulting molecule incorporates an electron poor dienophile and a reactive diene (the furan). Heat promotes the Diels-Alder reaction and delivers the target molecule.
Reference - Organic Letters 2006

Propose a mechanism for the following transformation.

- Answer
-
This mechanism combines steps from the Heck reaction, Suzuki reaction, and a carbonylation. The mechanism begins with an oxidative addition followed by an alkyne insertion (Heck reaction). There is no hydrogen on the beta carbon, so the reaction continues with CO association and insertion steps (carbonylation). At this point, transmetallation between Pd and B occurs followed by reductive elimination (Suzuki reaction) to yield the product. Predicting this as the product of the reaction would be difficult based on what we know. However, given the structure of the product, we do have the skills to propose a mechanism.

Contributors
- Prof. Kevin Shea (Smith College)