
15.8: Opening of Epoxides
- Page ID
- 45534
Epoxide ring-opening reactions - SN1 vs. SN2, regioselectivity, and stereoselectivity
The ring-opening reactions of epoxides provide an excellent review of the differences between SN1 and SN2 reactions. Both mechanisms are good examples of regioselective reactions. In a regioselective reaction, two (or more) different constitutional isomers are possible as products, but one is formed preferentially (or sometimes exclusively). Ring-opening reactions can proceed by either SN2 or SN1 mechanisms, depending on the nature of the epoxide and on the reaction conditions. For the SN1 mechanism, the stability of the charged intermediate determines the regioselectivity. For the concerted SN2 mechanism, sterics are the dominating consideration. If the epoxide is asymmetric, the structure of the product will vary according to which mechanism dominates. When an asymmetric epoxide undergoes solvolysis in basic methanol, ring-opening occurs by an SN2 mechanism, and the less substituted carbon is the site of nucleophilic reaction, leading to what we will refer to as product B:

Conversely, when solvolysis occurs in acidic methanol, the reaction occurs by a mechanism with substantial SN1 character, and the more substituted carbon is the site of reaction. As a result, product A predominates.

Let us examine the basic, SN2 case first. The leaving group is an alkoxide anion, because there is no acid available to protonate the oxygen prior to ring opening. An alkoxide is a poor leaving group, and thus the ring is unlikely to open without a 'push' from the nucleophile.
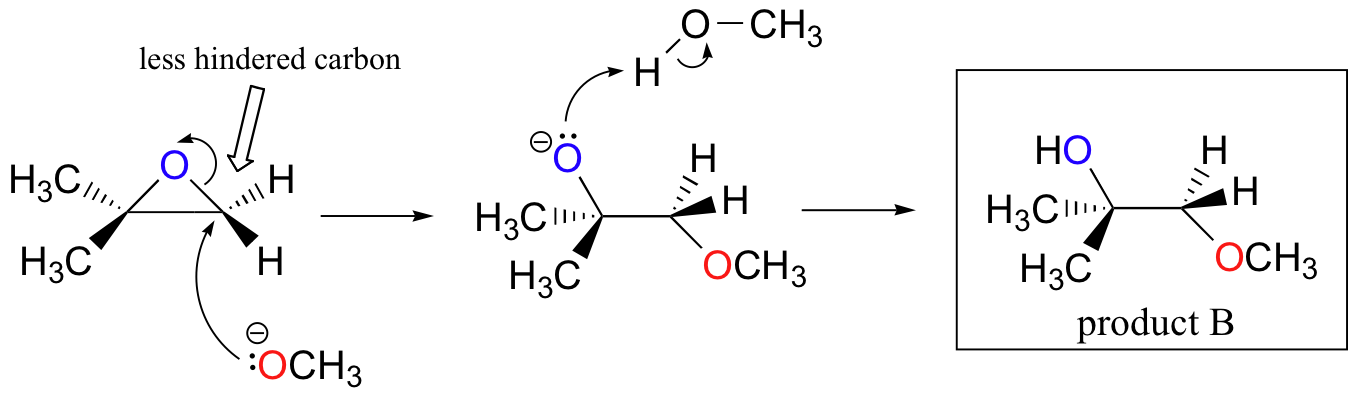
The nucleophile itself is a potent, deprotonated, negatively charged methoxide ion. When a nucleophilic substitution reaction involves a poor leaving group and a powerful nucleophile, it is very likely to proceed by an SN2 mechanism.
What about the electrophile? There are two electrophilic carbons in the epoxide, but the best target for the nucleophile in an SN2 reaction is the carbon that is least hindered. This accounts for the observed regiochemical outcome. Like in other SN2 reactions, bimolecular, nucleophilic substitution reactions take place from the backside, resulting in inversion at the electrophilic carbon.
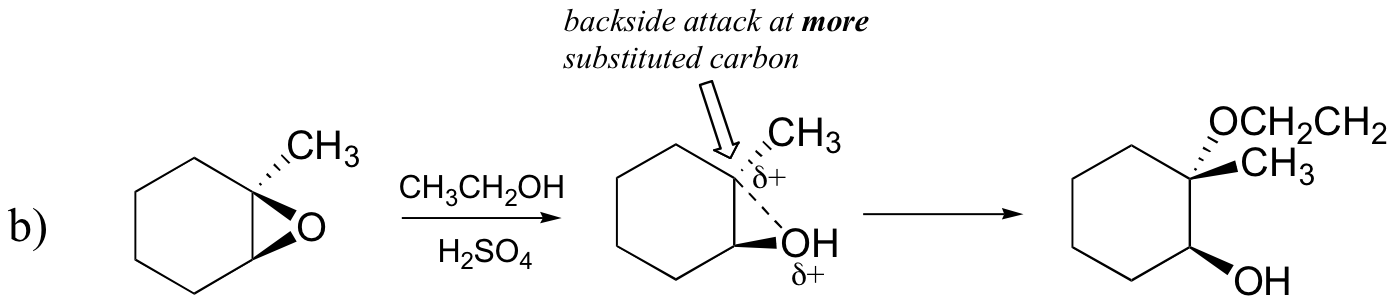
The acid-catalyzed epoxide ring-opening reaction mechanism is analogous to the formation of the bromonium ion in halogenation of alkenes and mercurium ion formation in oxymercuration/demercuratioin or alkoxymercuration/demercuration. First, the oxygen is protonated, creating a good leaving group (step 1 below) . Then the carbon-oxygen bond begins to break (step 2) and positive charge begins to build up on the more substituted carbon (recall carbocation stability).

Unlike in an SN2 reaction, the nucleophile reacts with the electrophilic carbon (step 3) before a complete carbocation intermediate has a chance to form.
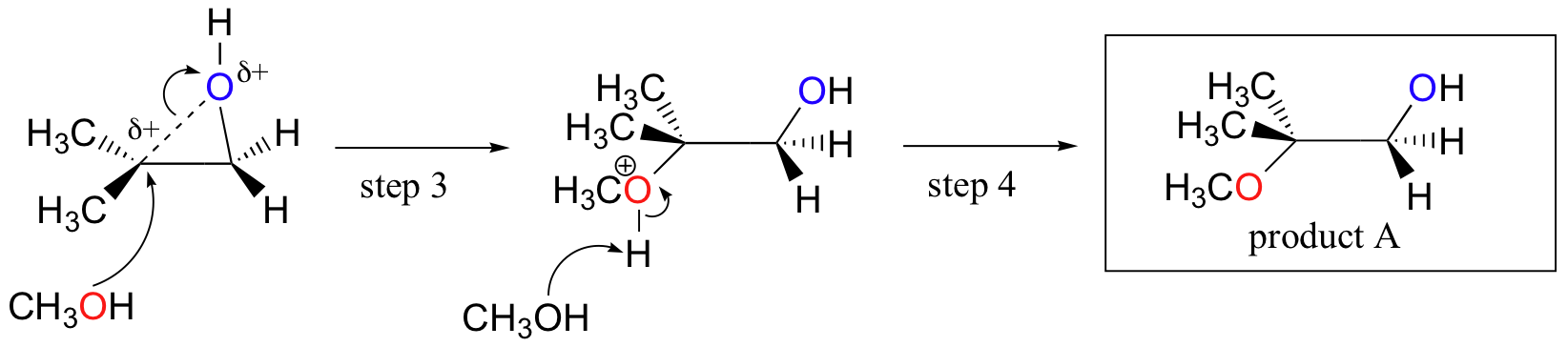
The reaction takes place preferentially from the backside (like in an SN2 reaction) because the carbon-oxygen bond is still to some degree in place, and the oxygen blocks reaction from the front side. Notice, however, how the regiochemical outcome is different from the base-catalyzed reaction: in the acid-catalyzed process, the nucleophile reacts with the more substituted carbon because this carbon that holds a greater degree of positive charge.
Example
Predict the major product(s) of the ring opening reaction that occurs when the epoxide shown below is treated with:
- ethanol and a small amount of sodium hydroxide
- ethanol and a small amount of sulfuric acid
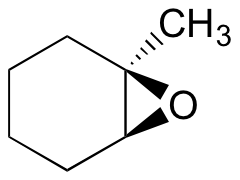
Hint: be sure to consider both regiochemistry and stereochemistry!
- Answer
-
Anti Dihydroxylation
Epoxides may be cleaved by aqueous acid to give glycols that are often diastereomeric with those prepared by the syn-hydroxylation reaction. Proton transfer from the acid catalyst generates the conjugate acid of the epoxide, which is attacked by nucleophiles such as water in the same way that the cyclic bromonium ion described above undergoes reaction. The result is anti-hydroxylation of the double bond. In the following equation this procedure is illustrated for a cis-disubstituted epoxide, which can be prepared from the corresponding cis-alkene. This hydration of an epoxide does not change the oxidation state of any atoms or groups.
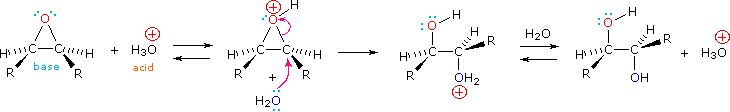
Addition of HX
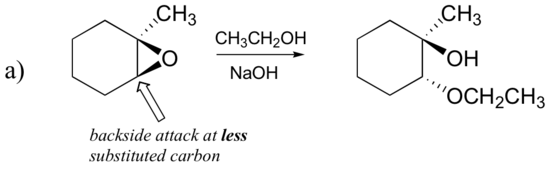
Epoxides can also be opened by other anhydrous acids (HX) to form a trans halohydrin. When both the epoxide carbons are either primary or secondary the halogen anion will react with the less substituted carbon and an SN2 like reaction. However, if one of the epoxide carbons is tertiary, the halogen anion will primarily react with the tertiary carbon in a SN1 like reaction.

Example
In the first example, the epoxide is formed by a secondary and primary carbon. The reaction with the Cl- nucleophile proceeds via the SN2 mechanism and reacts with the least substituted carbon.

In the second example, the epoxide is formed by a tertiary and primary carbon. The reaction with Cl- nucleophile proceeds via the SN1 like mechanism and reacts with the most substituted carbon because it carries the greater partial positive charge.

Exercise
9. Given the following, predict the product assuming only the epoxide is affected. (Remember stereochemistry)
10. Predict the product of the following, similar to above but a different nucleophile is used and not in acidic conditions. (Remember stereochemistry)
11. Epoxides are often very useful reagents to use in synthesis when the desired product is a single stereoisomer. If the following alkene were reacted with an oxyacid to form an epoxide, would the result be a enantiomerically pure? If not, what would it be?
- Answer
-
9.
Note that the stereochemistry has been inverted
10.
11.
First, look at the symmetry of the alkene. There is a mirror plane, shown here.
Then, think about the mechanism of epoxidation with an oxyacid, take for example mCPBA. The mechanism is concerted, so the original cis stereochemistry is not changed. This leads to "two" epoxides.
However, these two mirror images are actually identical due to the mirror plane of the cis geometry. It is a meso compound, so the final result is a single stereoisomer, but not a single enantiomer.
Contributors and Attributions
Dr. Dietmar Kennepohl FCIC (Professor of Chemistry, Athabasca University)
Prof. Steven Farmer (Sonoma State University)
William Reusch, Professor Emeritus (Michigan State U.), Virtual Textbook of Organic Chemistry