
7.6: Unimolecular Elimination: E1
- Page ID
- 32382
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\dsum}{\displaystyle\sum\limits} \)
\( \newcommand{\dint}{\displaystyle\int\limits} \)
\( \newcommand{\dlim}{\displaystyle\lim\limits} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\(\newcommand{\longvect}{\overrightarrow}\)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)Unimolecular Elimination (E1) is a reaction in which the removal of an HX substituent results in the formation of a double bond. It is similar to a unimolecular nucleophilic substitution reaction (SN1) in various ways. One being the formation of a carbocation intermediate. Also, the only rate determining (slow) step is the dissociation of the leaving group to form a carbocation, hence the name unimolecular. Thus, since these two reactions behave similarly, they compete against each other. Many times, both these reactions will occur simultaneously to form different products from a single reaction. However, one can be favored over another through thermodynamic control. Although Elimination entails two types of reactions, E1 and E2, we will focus mainly on E1 reactions with some reference to E2.
General Reaction
An E1 reaction involves the deprotonation of a hydrogen nearby (usually one carbon away, or the beta position) the carbocation resulting in the formation of an alkene product. In order to accomplish this, a Lewis base is required. For a simplified model, we’ll take B to be a Lewis base, and LG to be a halogen leaving group.
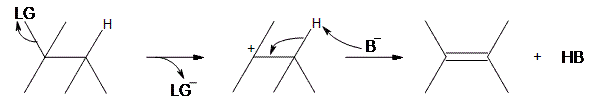
As can be seen above, the preliminary step is the leaving group (LG) leaving on its own. Because it takes the electrons in the bond along with it, the carbon that was attached to it loses its electron, making it a carbocation. Once it becomes a carbocation, a Lewis Base (\(B^-\)) deprotonates the intermediate carbocation at the beta position, which then donates its electrons to the neighboring C-C bond, forming a double bond. Unlike E2 reactions, which require the proton to be anti to the leaving group, E1 reactions only require a neighboring hydrogen. This is due to the fact that the leaving group has already left the molecule. The final product is an alkene along with the HB byproduct.
Reactivity
Due to the fact that E1 reactions create a carbocation intermediate, rules present in \(S_N1\) reactions still apply.
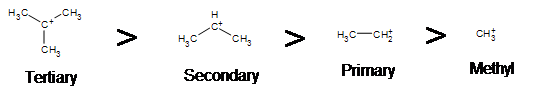
As expected, tertiary carbocations are favored over secondary, primary and methyl’s. This is due to the phenomena of hyperconjugation, which essentially allows a nearby C-C or C-H bond to interact with the p orbital of the carbon to bring the electrons down to a lower energy state. Thus, this has a stabilizing effect on the molecule as a whole. In general, primary and methyl carbocations do not proceed through the E1 pathway for this reason, unless there is a means of carbocation rearrangement to move the positive charge to a nearby carbon. Secondary and Tertiary carbons form more stable carbocations, thus this formation occurs quite rapidly.
Secondary carbocations can be subject to the E2 reaction pathway, but this generally occurs in the presence of a good / strong base. Adding a weak base to the reaction disfavors E2, essentially pushing towards the E1 pathway. In many instances, solvolysis occurs rather than using a base to deprotonate. This means heat is added to the solution, and the solvent itself deprotonates a hydrogen. The medium can effect the pathway of the reaction as well. Polar protic solvents may be used to hinder nucleophiles, thus disfavoring E2 / Sn2 from occurring.
Acid catalyzed dehydration of secondary / tertiary alcohols
We’ll take a look at a mechanism involving solvolysis during an E1 reaction of Propanol in Sulfuric Acid.

- Step 1: The OH group on the pentanol is hydrated by H2SO4. This allows the OH to become an H2O, which is a better leaving group.
- Step 2: Once the OH has been hydrated, the H2O molecule leaves, taking its electrons with it. This creates a carbocation intermediate on the attached carbon.
- Step 3: Another H2O molecule comes in to deprotonate the beta carbon, which then donates its electrons to the neighboring C-C bond. The carbons are rehybridized from sp3 to sp2, and thus a pi bond is formed between them.
Mechanism for Alkyl Halides
This mechanism is a common application of E1 reactions in the synthesis of an alkene.
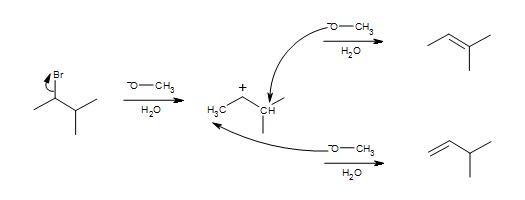
Once again, we see the basic 2 steps of the E1 mechanism.
- The leaving group leaves along with its electrons to form a carbocation intermediate.
- A base deprotonates a beta carbon to form a pi bond.
In this case we see a mixture of products rather than one discrete one. This is the case because the carbocation has two nearby carbons that are capable of being deprotonated, but that only one forms a major product (more stable).
How are Regiochemistry & Stereochemistry involved?
In terms of regiochemistry, Zaitsev's rule states that although more than one product can be formed during alkene synthesis, the more substituted alkene is the major product. This infers that the hydrogen on the most substituted carbon is the most probable to be deprotonated, thus allowing for the most substituted alkene to be formed.
Unlike E2 reactions, E1 is not stereospecific. Thus, a hydrogen is not required to be anti-periplanar to the leaving group.

In this mechanism, we can see two possible pathways for the reaction. One in which the methyl on the right is deprotonated, and another in which the CH2 on the left is deprotonated. Either one leads to a plausible resultant product, however, only one forms a major product. As stated by Zaitsev's rule, deprotonation of the most substituted carbon results in the most substituted alkene. This then becomes the most stable product due to hyperconjugation, and is also more common than the minor product.
Contributors
- Satish Balasubramanian
The E1 mechanism is nearly identical to the SN1 mechanism, differing only in the course of reaction taken by the carbocation intermediate. As shown by the following equations, a carbocation bearing beta-hydrogens may function either as a Lewis acid (electrophile), as it does in the SN1 reaction, or a Brønsted acid, as in the E1 reaction.
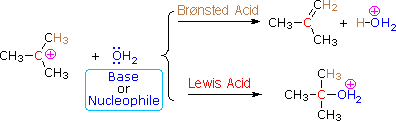
Thus, hydrolysis of tert-butyl chloride in a mixed solvent of water and acetonitrile gives a mixture of 2-methyl-2-propanol (60%) and 2-methylpropene (40%) at a rate independent of the water concentration. The alcohol is the product of an SN1 reaction and the alkene is the product of the E1 reaction. The characteristics of these two reaction mechanisms are similar, as expected. They both show first order kinetics; neither is much influenced by a change in the nucleophile/base; and both are relatively non-stereospecific.
(CH3)3C–Cl + H2O ——> [ (CH3)3C(+) ] + Cl(–) + H2O ——> (CH3)3C–OH + (CH3)2C=CH2 + HCl + H2O
To summarize, when carbocation intermediates are formed one can expect them to react further by one or more of the following modes:
- The cation may bond to a nucleophile to give a substitution product.
- The cation may transfer a beta-proton to a base, giving an alkene product.
- The cation may rearrange to a more stable carbocation, and then react by mode #1 or #2.
Since the SN1 and E1 reactions proceed via the same carbocation intermediate, the product ratios are difficult to control and both substitution and elimination usually take place.
Having discussed the many factors that influence nucleophilic substitution and elimination reactions of alkyl halides, we must now consider the practical problem of predicting the most likely outcome when a given alkyl halide is reacted with a given nucleophile. As we noted earlier, several variables must be considered, the most important being the structure of the alkyl group and the nature of the nucleophilic reactant. The nature of the halogen substituent on the alkyl halide is usually not very significant if it is Cl, Br or I. In cases where both SN2 and E2 reactions compete, chlorides generally give more elimination than do iodides, since the greater electronegativity of chlorine increases the acidity of beta-hydrogens. Indeed, although alkyl fluorides are relatively unreactive, when reactions with basic nucleophiles are forced, elimination occurs (note the high electronegativity of fluorine).
Contributors
- William Reusch, Professor Emeritus (Michigan State U.), Virtual Textbook of Organic Chemistry