
6.1: Colchicine
- Page ID
- 285463

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\dsum}{\displaystyle\sum\limits} \)
\( \newcommand{\dint}{\displaystyle\int\limits} \)
\( \newcommand{\dlim}{\displaystyle\lim\limits} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\(\newcommand{\longvect}{\overrightarrow}\)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)Biosynthetic Strategy
The use of aromatic amino acids, i.e. phenylalanine or tyrosine, as the sole starting materials for colchicine (8) requires preparing the seven-membered C-ring by expansion of an aromatic six-membered ring in a precursor such as 9. How the ring expansion is accomplished will considered later. Since the starting amino acids do not have benzylic amino groups, it is likely that the bond between the C ring and the benzylic carbon is formed by electrophilic aromatic substitution. Since the starting amino acids are aryl propionic acid derivatives, the electrophile could be derived from an aryl propionic amide such as 10. Clearly the bond between the A and C rings in 10 would be formed by oxidative coupling of electron rich aryl precursors 11 and 12. Since the nitrogen in 11 probably comes from an amino group in the C ring amino acid precursor, and since 12 should also be derived from an a-amino arylpropionic acid, it is likely that the structure 12 must be revised so that the R group in 11 incorporates 12 as in the amide 13 (see below). A cinnamic acid 14, generated by elimination of ammonia from phenylalanine (6) could be the progenitor of the arylpropionic acid portion of the amide 13.

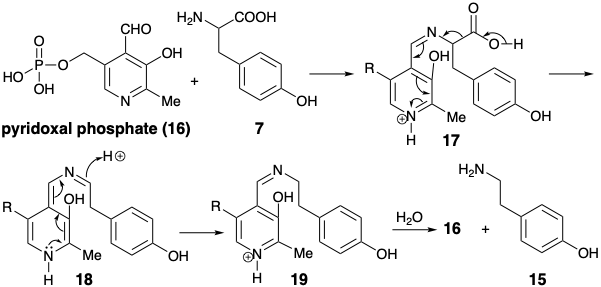
Generation of a phenylethyl amine 15 by decarboxylation of tyrosine (7) requires cleavage of a bond lying on a dissonant circuit. Such a cleavage is achieved biosynthetically by a polar process which converts the amino group temporarily into a derivative that can stabilize electronic excess on the amino carbon. The decarboxylation of tyrosine (7) to give p-hydroxy-b-phenthyl amine (15) is an example of a general reaction of amino acids that is promoted by the coenzyme pyridoxal phosphate (16). The process is initiated by the formation of a Schiff base 17. The pyridine nitrogen is conjugated with the carbon a to the carboxyl and can stabilize electronic excess at that carbon. The imine nitrogen does not provide polar activation; it serves merely as a linking atom. Schiff base 17 readily undergoes acid catalyzed decarboxylation to form 18. Protonation of 181 leads to rearomatization delivering the Schiff base 19. Hydrolysis delivers the phenylethyl amine 15 and regenerates 16. Thus, pyridoxal phosphate (16) acts as a polar reactivity-inverting catalyst.
We encountered polar ring expansion strategies in the total synthesis of longifolene (see section 4.4). There, a fused bicyclic intermediate, generated by cyclopropanation of a cyclohexene, underwent cleavage of the fusion bond in conjunction with departure of a nucleofuge to deliver a cycloheptenyl array (eq 1). An analogous process, involving departure of an electrofuge in conjunction with cleavage of the fusion bond in a similar intermediate, depends on a cyclopropyl carbinyl to homoallyl carbocation rearrangement (eq 2). The presence of oxygen substituents in the colchicine C ring suggests the possibility that such a ring expansion mechanism may transpire during the biosynthetic conversion of a six-membered C ring precursor into the seven-membered C ring.

One carbon of the phenethyl sidechain in 13, the benzylic carbon, could be incorporated in an aromatic precursor to generate the seven-membered C ring. The remaining carbon of the sidechain must be disconnected. Disconnection of this carbon as a carbocation can be stabilized by the amino group as in 20. Retrosynthetic analysis of the colchicine biosynthetic ring expansion, presuming 20 as electrofuge, suggests cyclopropyl carbinyl and homoallyl carbocation intermediates 21 and 22. The carbon that is inserted into the aromatic ring of the starting material 24 corresponds to the dication synthon 23 for which an aldehyde might serve as a synthetic equivalent.

Biosynthesis
In the biosynthesis of colchicine (8), \(\ce{NH3}\) is eliminated from phenylalanine (6), and tyrosine (7) is decarboxylated, before an amide 26 is formed by joining the resulting intermediates 25 and 15. The seven-membered B ring is created by an enantioselective intramolecular electrophilic aromatic substitution that gives 27 and an oxidative coupling of 28 that delivers 29. Functionalization of an apparently unactivated methylene in 29 is accomplished by a sequence involving polar hydrolytic fragmentation to 30 followed by oxidation to an aldehyde suitable for insertion into the six-membered C ring progenitor in 31. Expansion of the aryl ring to generate a seven-membered tropolone is initiated by intramolecular electrophilic aromatic substitution. Intramolecular alkylation of 32 followed by a fragmentation of an intermediate cyclopropane 33 produces the ring expanded skeleton in 34 of the biosynthetic target. Final hydrolysis of the imminium group, N-demethylation and N-acetylation delivers colchicine (8).

Molecular Characteristics
The stability of aromatic derivatives is often exploited in synthesis by strategies that incorporate preformed aromatic moieties. Thus, in the biosynthesis of colchicine (8), the aromatic A-ring is derived from the preformed aromatic ring of phenylalanine (6). The four different total syntheses of colchicine to be considered in this section all adopt this same strategy. In contrast with the biosynthesis, however, the total syntheses all employ fully functionalized aromatic starting materials. This is because the regioselective hydroxylations, that are acheived enzymatically in the biosynthesis, are not so readily achieved in the laboratory.
It should also be noted that the C ring of colchicine (8) contains two functional groups that provide electrophilic activation on adjacent carbon atoms, a polar reactivity dissonance. Thus, these functional groups cannot be exploited directly in a polar reaction (i.e. without umpölung) to create the C-C bond of the dissonant circuit between these carbon atoms. In each of the following syntheses, the seven- membered C ring is added to an AB-ring precursor. In each case, a different strategy is exploited for annelation of the C ring.
Key Intermediate-Directed Strategies for Conchicine
Two syntheses of colchicine exploited a readily available key intermediate, purpurogallin (34), for the AB ring moiety of 8 and formed the seven-membered C-ring by nonpolar reactions. Polar reactivity analysis of 34 suggests a synthesis from an aromatic precursor 36. Thus, appendage to 36 of the B-ring in 34 can be facilitated by addition of an activating carboxyl as in 35 that could be generated from 36 and 37 by two polar bond-forming reactions. In fact, the aromatic starting material 36 can also be the precursor of a temporarily-bridged synthetic equivalent 39 (vide infra) of the synthon 37.

Purpurogallin (34) was a well-known product from the oxidation of pyrogallol (36). It is probably formed by dimerization of 3-hydroxy-o-quinone (38). An initial Michael addition of the enolate 39 to 38 to give 40 followed by an intramolecular aldol condensation leads to a tricyclic intermediate 41 (see below). This is cleaved in a retro Dieckman reaction, typical for β-diketones, to produce a bicyclic carboxylic acid 37, which can be isolated. This vinylogous β-keto acid readily undergoes decarboxylation to deliver 28.


Both syntheses employing 34 for the A and B rings simplified the target by neglecting the acetamido group. This could be introduced by benzylic oxidation after completion of the carbon skeleton of the target 8. Both syntheses construct the simplified target 43 from benzosuberone derivative 44, in which the carbonyl functional group provides activation for annelation of the C ring. Eschenmosher1 prepared 39a by reducing the trimethyl ether 45 of purpurogallin (34) via 46, 47, and 48.

A Cycloaddition-Pericyclic Rearrangement Strategy for the C Ring
Eschenmosher's strategy1 for annelation of the seven membered ring C was to build a diene onto 44a, then construct a six membered carbocycle by a Diels-Alder reaction of the diene and finally expand the six to a seven membered ring by pericyclic rearrangement of a norcaradiene. Facile interconversion of cycloheptatrienes such as 49 with norcaradienes such as 50 is a well-known [3.3] sigmatropic (Cope) rearrangement that is driven to favor cycloheptatrienes by the relief of ring strain associated with cleavage of the cyclopropane. It is doubtful that the strategy was concieved by rigorous retrosynthetic analysis since conversion of 49 to 43 would certainly require extensive functional group manipulations. The decision to employ 49 as a precursor for 43 almost certainly evolved as a consequence of the decisions to employ: (1) a Cope rearrangement of a norcaradiene.

Therefore, chloromethylmaleic anhydride (52) is ideally suited to cycloadd to the reletively electron rich diene 53. Perhaps a more obvious dislocation of 50 would be to 53 and the cyclopropene 55. This branch of the retrosynthetic tree would, most probably, be considered first because it would provide a more convergent synthesis. Thus, reaction of 55 with α-pyrone 53 would deliver 50 directly by a Diels alder cycloaddition, followed in situ by a retro Diels Alder cycloelimination of carbon dioxide from an intermediate 56.

The alternative precursor, dienophile 52 with a chloromethyl substituent, is suggested by polar disconnection of 50 to 51 that can be derived from 53 by a Diels-Alder-retro Diels-Alder sequence. Dienophile 52 rather than 55 was chosen because it is more readily available than 55. A Diels-Alder addition of 52 to 53 followed by a retro Diels-Alder elimination of \(\ce{CO2}\) from an intermediate adduct, a carbonyl-alkene exchange process, will provide the cyclohexadiene 51. The driving force for the process is the generation of a relatively stable C=O bond in exchange for the C=C bond of the dienophile 52. The diene 53 is an enol lactone derivable from the acid 54. Polar analysis of 54 suggests construction from 44a and methyl propiolate by a polar 1,4-addition. Apparent Michael alkylation of 44a with methyl propiolate provided pyrone 57. Though 44a could give 57 via direct Michael addition to the yneone, in fact the reaction was more complex. It involved participation by the phenolate anion. Thus, the electrophile was delivered intramolecularly to the rather hindered benzylic carbon of 44a.

After methylation of phenol 57, the annelation of a cyclohexadiene 58 was achieved by the well- known cycloaddition-cycloelimination reaction of α-pyrones. Base-catalyzed intramolecular alkylation of the diester 51 from 58 led, via the norcaradiene 50, to the cycloheptatriene 49. The least hindered ester in 49 was readily hydrolyzed selectively, and the resulting acid afforded a tropolone 61 via osmium catalyzed vicinal hydroxylation-decarboxylation, saponification of the remaining ester in 60 and a second decarboxylation.

Unfortunately, the sequence leads to an oxygen function on C-11 rather than C-9 as required for colchicine. Transposition of the functional group was achieved by a well-precedented sequence of nucleophilic displacements on the tosylate derivative 62 of 61 first with \(\ce{NH3}\) to give 63 then with -OH to deliver 64. The tropolone 64 was then methylated and functionalized at C-7 by allylic bromination with N-bromosuccinimide to provide 65. Nucleophilic displace-ment of bromide by ammonia gave the required C-7 amine accompanied by ammonolysis of the tropolone, a vinylogous ester. Saponification of the resulting vinylogous amide 66 gave 67, that afforded colchicine upon methylation and acetylation.

An Acyloin Strategy for the C Ring
Van Tamelen's strategy for annelation of the C-ring with vicinal oxygen functionality recognizes the applicability of an intramolecular acyloin reaction for creation of the dissonant circuit between vicinal electrophilic activating groups.2 Further polar analysis suggests a synthesis of the requisite diester intermediate 68 by exploiting the polar activation provided by a carbonyl group in an AB-ring precursor 44. Thus, appendage of acetic and a propionic acid side chains should be feasible respectively by a Reformatsky reaction and Michael alkylation.

A pair of isomeric hydroxy diacids 69c and 69t was obtained by Michael alkylation of 44b with acrylonitrile, Reformatsky reaction of the intermediate ketonitrile and hydrolysis. The hydroxyl group was masked intramolecularly as a lactone, and the remaining carboxyl group was methylated. Only one of the isomeric lactone esters 70 underwent an acyloin reaction which provided 71. Unfortunately, this was the minor isomer 70t, with an axial carbometh-oxymethyl substituent. The ester groups in the major isomer 70c could not readily attain juxtaposition suitable for intramolecular acyloin reaction. The acyloin product 71 was oxidized to 72 with Cu(II) and further oxidized with NBS to provide tropolone 64. Methylation and bromination delivered the bromide 65, an intermediate also prepared by Eschenmosher. Substitution of an amino group for the bromo substituent in 65 followed by hydrolysis, remethylation of the vinylogous carboxylic acid, and N-acetylation produced colchicine 8.

Extensive functional group manipulations after completion of the carbon skeleton were required in the Eschenmosher synthesis because an annelation strategy for the C ring was adopted that ignored target related functionality. Van Tamelen could complete his synthesis with less functional group manipulations because more target related functionality, that had been exploited to facilitate skeletal construction, was present after completion of the C ring.
A Target-related Functionality-promoted Polar Bond Formation Strategy
Another synthesis of colchicine (8), also involving annelation of ring C on a preformed AB-ring intermediate, was devised by R. B. Woodward.3 The strategy is unique in incorporating the 7-amino substituent early and in its extensive use of target related functionality to facilitate carbon skeletal construction. Dislocation of the target 8 to derivative 74 in which the 8-position is also blocked allows selective introduction of oxygen at C-10. In the synthetic equivalent 75 of the synthon 74, an aromatic isothiazole ring masks both the amino substituent and C-8. Annelation of the C-ring can be achieved by a Dieckman cyclization 77 → 76 exploiting the polar activation provided by the C-9 carbonyl and an activating carbomethoxyl group appended to C-10 in a precursor 77. The polar activation provided by this carbomethyl group in 77 suggests an electrophilic aromatic substitution 78 → 77 for annelation of the B- ring. Of course, for the appropriate electrophilicity to be expressed, the carbomethoxyl group in 77 must be conjugated with the γ-position as in 78. The isothiazole also serves as a temporary bridge in 78, that assists entropically in the 78 → 77 cyclization. The carbomethoxyl in 78 also allows a polar elaboration of the dienoic ester array from an isothiazole aldehyde precursor 79 by stabilizing a carbanion in the ylide fragment 80. The sulfur atom in the isothiazole ring even provides activation for carbanion generation a to sulfur in 78 allowing polar connection of the carbomethoxyl required in 77. The extensive strategic utilization of the isothiazole unit in Woodward's strategy is a hallmark of this synthetic plan.

After Dieckman cyclization of 81, the C-9 monoketone 75 is selectively oxidized at the C-10 by a polar reaction exploiting the nucleophilic activation at C-10 provided by the C-9 carbonyl group. Nucleophilic attack by the C-10 carbanion on a sulfur electrophile leads to oxidation of C-10 (and concomitant reduction of sulfur). The resulting thioketal 82 is hydrolyzed to 83, that affords the enol acetylation product 84. The enediolate obtained by saponification of the diacetate 84 is readily oxidized to deliver 85. Desulfurization of 85 with Raney nickel removes the isothiazole masking group. Reduction of the resulting imine 86 followed by acetylation provides 87 that is N-methyl-ated to deliver colchicine (8).

Woodward's synthesis of the key intermediate 81 is centered around the novel aromatic isothiazole ring. The starting material, an isothiazole 88, is readily available from methyl β-aminocrotonate, an enamine derived from methyl acetoacetate. The conjugated methyl group in 88 is readily brominated with NBS. Alkylation of \(\ce{Ph3P}\) affords a phosphonium bromide 89, that gives ylide 90 upon deprotonation. Wittig olefination of 3,4,5-trimethoxybenzaldehyde (91) with ylide 90 produces alkene 92 that was selectively hydrogenated with diimide. Catalytic hydrogenation of 92 was precluded by the susceptibility of the isothiazole ring to hydrogenolysis. Hydride reduction of the ester 93 and partial oxidation of an intermediate conjugated carbinol gave the aldehyde 94. Wittig olefination of 94 with ylide 95 followed by saponification and cis-trans isomerization provided 96.

This diene underwent intramolecular electrophilic aromatic substitution upon treatment with perchloric acid. The temporary bridge provided by the isothiazole ring undoubtedly facilitates this cyclization by favorably juxtaposing the reacting carbon centers. Selective reduction of the cyclization product 99 with diimide gave 100. The final carbon required for the C ring was introduced by carbonylation of an organolithium derivative 102 from the thiazole 100. Thus, selective metallation in the presence of a carboxylate was achieved with the relatively non nucleophilic, sterically encumbered aryl lithium 101. The lithiated thiazole 102 gave the key intermediate 81 upon carbonation followed by methylation.

A Hypothetically Biomimetic Strategy
A fourth synthesis of colchicine, devised by Scott4, differs from the previous three in its strategy for skeletal constuction. Scott assembled an intermediate containing the A and C rings and then created the B ring by an intramolecular oxidative coupling. This strategy was based on a hypothetical mechanism for colchicine biosynthesis, that is now know not to be operative. In this mechanism, in contrast with the actual biosynthetic mechanism (see above), generation of a tropolone C-ring by ring expansion of an aromatic precursor preceeds the oxidative coupling that creates the B-ring.

The tactic of introducing the colchicine amino group at the end of the synthesis was well-precedented. Thus, the Scott strategy begins with dislocation of the target 8 to a precursor 103. The bond between aryl and tropolone rings lies along a dissonant circuit between the oxygen functionality at positions 1 + 9, 1 + 10, 3 + 9, or 3 + 10. Thus, polar analysis reveals that formation of this bond cannot be achieved by a polar process using these functional groups to provide activation since union of two nucleophilic centers would be required. Such a process can be achieved oxidatively, suggesting dislocation of 103 to a precursor 104 with two monocyclics joined by a simple trimethylene bridge. The polar reactivity afforded by the carbonyl group in 104 can be reinforced by activating carboxyl groups in a precursor 105 allowing polar bond formation between an electrophilic intermediate 106 and a nucleophile derived from 107 by deprotonation. The unobvious choice of 107 as a precursor for 104 was undoubtedly dictated by its ready availability from purpurogallin 34 by selective oxidative cleavage of the relatively electron rich aryl ring.

Thus, oxidation of 34 with hydrogen peroxide followed by dehydration gave the enol anhydride 107. It is interesting that Scott utilized purpurogallin (34) as a precursor for the C ring of colchicine in contrast to Eschenmosher and Van Tamelen who built the A and B rings of colchicine from 34.

An A-ring synthon, 3,4,5-trimethoxy acetaldehyde (106) was obtained by Arndt-Eistert homologation of 3,4,5-trihydroxybenzoic acid (108). Union of the electrophilic A ring synthon 106 with the nucleophilic C-ring synthon 107 was best achieved thermally without base catalysis by a process that starts with an aldol condensation. At 100°C, a lactone 110 was formed, presumably by decarboxylation of an intermediate vinylogous β-keto acid 109. Further heating of 110 at 190°C gave 112, presumably by decarboxylation of an intermediate β-keto acid 111. Reduction and demethylation gave the A+C ring intermediate 104. The electron rich product 103 from the desired oxidative coupling of 104 was highly susceptible to undesirable further oxidation. Nevertheless, the mild oxidant, \(\ce{FeCl3}\), in a two phase system gave 103 after paper chromatography in an inert atmosphere albeit in low yield (5%).

It has been suggested that the annelation may involve an ionic process rather than the radical coupling originally envisioned.5 Thus, utilizing the aryl oxygen functionality at position 2, that was ignored in the polar analysis of 103 above, it can be seen that the bond between the aryl and tropolone rings lies on a consonant circuit between positions 2 and 10. This allows Michael addition of a tropolone nucleophile to an enone electrophile as shown in 113 to deliver 114. Final adjustment of functionality gave 8 from 103 through 73 as discussed previously.
