
5.4: Erythronolide B
- Page ID
- 285459

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\dsum}{\displaystyle\sum\limits} \)
\( \newcommand{\dint}{\displaystyle\int\limits} \)
\( \newcommand{\dlim}{\displaystyle\lim\limits} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\(\newcommand{\longvect}{\overrightarrow}\)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)An enzyme promoted reaction of propionylCoA with methylmalonylCoA generates 83 enantioselectively. Diastereoselective reduction then delivers the b-hydroxy thioester 84. In contrast with fatty acid biosynthesis, dehydration and conjugate reduction of the resulting α,β-unsaturated thioester does not preceed Claisen condensation with a second equivalent of methylmalonylCoA during the biosynthesis of the seco acid precursor 87 of the macrolide 6-deoxyerythronolide B.

The resulting β-ketothioester 85 is reduced to a β-hydroxyester 86 that reacts with additional methylmalonylCoA and NADPH to generate 87. Lactonization then provides 6-deoxyerythronolide B from which erythronolide is formed by oxidation. Generation of only 1 of the 2048 possible diastereomers of the acyclic intermediate 87 is the remarkable consequence of the asymmetry of the homochiral catalysts (enzymes) that promote the condensations and reductions responsible for producing ten asymmetric centers.

A Relay-directed Strategy for Total Synthesis of Erythronolide B.
In a feasibility study, naturally derived erythronolide B was converted into an acyclic hydroxy acid 88 that was used to work out the final steps for a total synthesis.12 The synthetic design process was then channeled by the choice of this naturally derived precursor, a relay compound. The two stereocenters at positions 12 and 13 in 88 are remote from those at positions 2-6 and 8. Therefore, it would be difficult to generate one set of stereocenters under the stereo-controlling influence of the other. Rather, building blocks containing these stereocenters with the correct absolute configurations can be assembled and then united to provide the relay compound 88. The first total synthesis of Erythronolide B1 3 adopted a convergent absolute asymmetric (see section 4.6) strategy designed to provide 88 by the union of two homochiral segments, a nucleophile 89 and an electrophile 90.

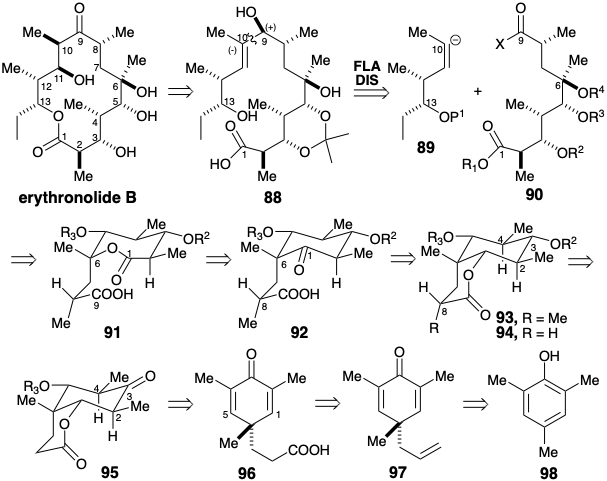
To provide a more conformationaly rigid platform for generating and confirming the relative configurations of stereocenters, a cyclic temporarily-bridged precursor 91 was envisioned for the acyclic subtarget 90. Even greater rigidity is provided by the smaller ring of a six-membered ketone 92 that incorporates the lactone functionality in latent form. Stereoelectronically favored axial delivery of hydrogen to the isomeric cyclohexanone 94 can be expected to generate an equatorial hydroxyl at position 3. Furthermore, dislocation of 93 to 94 allows a carbonyl at position 3 to promote a thermodynamically favored equatorial disposition for the methyls at positions 2 and 4, and sets the stage for uncovering a symmetrical precursor, vide infra. Another temporary bridge, a six-membered lactone can be exploited to favor generation of the required configuration at the relatively remote stereocenter at position 8 in 92 during a methylation of 95 that can be expected to favor an equatorial methyl in 94. Lactone bridges can also be exploited to assure the proper stereo and regiochemical orientation during introduction of oxygen substituents at positions 1 and 6 by polar additions to the C=C bonds in a symmetrical dienone precursor 96. A search of the literature for available starting materials with the carbon skeleton of 96 can be used to identify the allyl cyclohexadienone 97 that is readily prepared from the trimethylphenol 98.
Reaction with allyl bromide of a phenolate from 98 provides 100 by Claisen rearrangement of the initial O-allylation product 99. An acid-catalyzed Cope rearrangement of 100 delivers the symmetrical dienone 97 that is selectively hydroborated at the terminal vinyl group and then oxidized to produce the carboxylic acid 96. Stereo and regioselective delivery of oxygen to position 5 is accomplished by intramolecular addition of a carboxylate to a C=C bond in 96. Capture of the reversibly formed enolate carbanion intermediate with electrophilic bromine produces 101. To repeat this process on the remaining C=C bond, the lactone is saponified to regenerate a carboxylic acid. This also generates an epoxide by intramolecular displacement of the bromo substituent by alkoxide. A second stereo and regioselective delivery of oxygen, this time to position 1, is again accomplished by intramolecular addition of a carboxylate to a C=C bond. Subsequent reductive removal of unneeded heteroatom functionality at positions 1 and 4 provides 103.

The nucleophilic activation afforded by the lactone carbonyl in 103 could now be exploited to introduce a methyl group at position 8. However, prior adjustment of functionality level at position 3 avoids nucleophilic activation adjacent to the ketone carbonyl. Methylation of 104 then afforded 105 stereoselectively. The lactone in 103 differentiates the hydroxyl substituents at positions 1 and 5. To maintain this diferentiation after saponification of the lactone, the other hydroxyls in 105 had to be suitably masked. The choice of benzoate ester masking group is particularly subtle. The feasibility of selective saponification of the lactone in the presence of benzoate esters relies upon the diminished electrophilicity of the benzoate carbonyl group owing to conjugation. Oxidation of the alcohol 106 to a ketone and then a lactone 107 followed by activation of the carboxylic acid as a thioester provided an electrophilic C1-9 segment equivalent to 90 where the masking groups R1 and R4 are replaced by a lactone bridge.
The synthesis outlined above provides racemic compounds. Resolution of an early intermediate, the carboxylic acid 102, by fractional crystallization of diastereomeric 1-α-naphthylethylamine salts, was employed to generate intermediates with the absolute configurations shown above that are required for natural erythronolide B.
Resolution of an early intermediate was also employed to prepare the requisite enantiomer of a precursor 115 (see section 6.3) for the nucleophile 89. Thus, the epoxy carboxylic acid 110, that is readily available by a one-step oxidation of trans-crotyl alcohol (109), was resolved by fractional crystallization of diastereomeric 1-α-naphthylethylamine salts. Stereospecific nucleophilic substitution on 110 generated the absolute configuration required at position 12. The regioselectivity of this epoxide opening is controlled by the bulky ether substituent in 111. Use of the corresponding epoxy alcohol in the displacement showed much inferior regioselectivity. Replacement of the terminal hydroxyl in 112 by a methyl group to give 113 could be accomplished without masking of the secondary hydroxyl by using an excess of \(\ce{Me2CuLi}\). The completely regioselective conversion of acetylene 114 into vinyl iodide 115 depended upon the outstandingly high regioselectivity that had recently been reported for hydrozirconation of unsymmetrically disubstituted acetylenes. This step in the Corey erythronolide B synthesis is a poignent example of the impact of developments in synthetic methodology on our ability to achieve efficient syntheses of complex organic molecules.

Although both building blocks 108 and 115 were available in homochiral form with the correct absolute configurations for erythronolide B, the synthesis was actually carried out by coupling the correct enantiomer of 115 with racemic 108. Thus, a Grignard reagent 116 derived from 115 was acylated with thioester 108 to produce ketone 117 and a diastereomer in 90% total yield. The mixture was caried through several additional steps before separation by preparative thin layer chromatography.

Thus, reduction of the ketone carbonyl in 117 proved unexpectedly difficult owing to suprisingly similar reactivity of the keto and lactone carbonyls toward most reducing agents and also because of a proclivity toward conjugate reduction of the enone. Reduction with zinc borohydride was accompanied by two unexpected phenomena, a very welcomed complete stereoselectivity and an essentially irrelevant translactonization that generated a 10-membered lactone 118 after removal of the silyl protecting group at position 13. Saponification of this lactone was most effectively accomplished with \(\ce{LiOH}\) and aqueous \(\ce{H2O2}\) which presumably benefits from the supernucleophilicity of the hydroperoxide anion. Hydrolysis of the less reactive benzoate esters in 119 was then accomplished with aqueous \(\ce{KOH}\). Subsequent methyl- ation delivered 120 together with a diastereomer from which it was sepatated by TLC on silica gel.

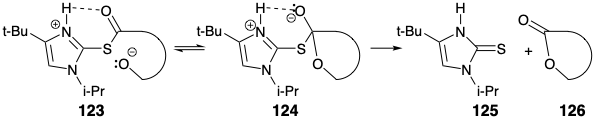
The relay compound 88 was obtained from 120 by ketalization with 2-methoxypropene, selective hydrolysis of 2-methoxy-2-propyl ethers that were also formed, and saponification of the methyl ester. Macrolactonization was accomplished by the "double activation method" that involves simultaneous activation of the hydroxyl and carboxyl functions. Presumably, a doubly activated intermediate 123 collapses to a tetrahedral carbonyl adduct 124 from which the lactone 126 is formed by elimination of 125. Thus, heating the thioester 122 at reflux in dry toluene provided erythrololide B in 50% yield.
Erythronolide B from Sugar-derived Homochiral Building Blocks.
A strategy for an enantiospecific total synthesis of erythronolide B evolved from the recognition that the C2-4 and C10-12 segments are identically substituted but have different absolute stereochemistries. Such segments, differentially substituted at each end, i.e. 127 and 128, might be elaborated and joined to generate the natural product. Therefore, studies were launched to define synthetic routes to such intermediates. Since (R)-2,3-O-isopropylideneglyceraldehyde (129) is a readily available homochiral building block (see section 3.7), it's possible utility as a starting material for the enantiospecific synthesis of such segments was explored.14

The addition of a crotylchromium reagent to aldehyde 129 showed virtually no diastereofacial selectivity for addition to the aldehyde but a high preference for generating an anti relationship at the two newly formed stereocenters owing to a stereoelectronic preference for chair-like transition state structures 130 and 131 that lead to 132 and 133. These diastereomers were readily separable by preparative column chromatography on a large scale. Conversion of 132 to an intermediate of type 127 and of 133 to an inter- mediate of type 128 requires inversion of the free secondary hydroxyl and substitution of the other secondary oxygen substituent by methyl with inversion of configuration. Inversion of the free hydroxyl was accomplished by activation as a tosylate followed by intramolecular SN2 displacement by a vicinal hydroxyl. This stereospecifically produced 135 from 134 and 138 from 137. Reaction of these epoxides with \(\ce{Li2Me2CuCN}\) accomplished the second configurational inversion during replacement of an oxygen substituent with methyl. The diols 136 and 139 correspond to the fragments 127 and 128 respectively, where FG1 and FG3 are both latent aldehydes while FG2 and FG4 are both hydroxymethyl groups.

The availability of the homochiral building blocks 136 and 139 channeled a second phase of strategic planning.15 Polar disconnection of erythronolide B at the C6-C7 and C8-C9 generates two precursors, 140 and 142, both with terminal carbonyl functions. Polar union of these fragments would require a "vicinally dianionic two-carbon (C6/C7) synthon" 141 with a pendant methyl group. Although the identification of a synthetic equivalent for 141 was postponed, it was recognized that "the methyl branching excluded the straightforward application of some acetylenic derivative."
Further polar disconnection of 142 to generate an aldehyde 143, that should be available from the building block 139, requires an acetyl carbanion synthon for which isopropenylmagnesium bromide is a latent synthetic equivalent. The alkene 144 is a latent equivalent of aldehyde 140. Further polar dislocation of 144 suggests an ethyl nucleophile and aldehyde electrophile 145 that should be available by oxidation of 136.

A masked derivative 147 of 142, containing the carboxylic acid functionality in latent form as a benzyloxy ether, was prepared from the homochiral building block 139 (see below). The addition of a Grignard reagent to an aldehyde intermediate generated the stereocenter at position 5 nonstereoselectively, leading to 146 as a 2:1 mixture of diastereomers. However, the requisite configuration at this carbon could be established by equilibration of the epimeric ketones 147 and 148. The equatorial ketone was favored over the axial epimer 148 by 94:6 at equilibrium.
A masked derivative 149 of aldehyde 140 was prepared from the homochiral building block 136. Although a polar union of the two carbonyl containing fragments 147 and 149 might exploit a dissonant dianionic fragment corresponding to 141, a synthetic equivalent of 141 was not devised. Rather, a nucleophilic reagent that was nucleophilic at C6 and contained an electrophilic carbon at position 7 was joined with aldehyde 151, and then the polar reactivity of C7 was inverted by conversion to an allylic thioether that could be deprotonated to provide nucleophilic reactivity at C7.

The carbon skeleton of erythronolide B was completed by joining ketone 147 with the allylic carbanion 152 produced by deprotonation of sulfide 151 with n-BuLi in the presence of TMEDA. Initial results were disapointing because the major product was the γ-adduct 153 rather than the desired α-adduct 154. By addition of HMPA, the formation of 153 could be suppressed almost completely. However, under these conditions, the main product was an epimeric α-adduct 155. Finally, it was discovered that precomplexation of the ketone 147 with \(\ce{BF3}\) strongly favored the required regio and stereoselecivity.

Having served its purpose as a polar reactivity inversion operator, the allylic phenylthio substituent was removed reductively. Differentiation of the hydroxyl groups was then accomplished by acetylation followed by selective deacetylation of 156 to unmask the primary hydroxyl. Oxidation followed by exhaustive deacetylation delivered the trihydroxy acid 157. Macrolactonization was accomp- lished in very good yield by conversion to a mixed anhydride that was cyclized in dilute toluene solution. Apparently a strain-free conformation that is ideally suited for cyclization is available to 157, whereas serious congestion is present in conformations suitable for forming a 12-membered lactone by acylation of the 11-OH.

Completion of the synthesis required introduction of oxygen at position 9. Anti Markovnikov hydration of the 8,9-C=C bond in 158 by hydroboration-oxidation accomplished this functionalization, and apparently owing to macrocyclic conformational effects, generation of the correct configuration at position 8 was favored by 9:1. Conformational effects also fostered selective oxidation of the secondary hydroxyl at position 9 in the presence of another secondary hydroxyl at position 11. Thus, the accumulation of many favorably selective steps owing to subtle, unanticipated consequences of molecular shape -- i. e. the remarkably effective macrolactonization, and favorably stereo and regioselective processes - - resulted in a total synthesis that rivals the Corey strategy that was more meticulously planned by thorough reterosynthetic analysis.
