
4.4: Syntheses of Longifolene
- Page ID
- 285452

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\dsum}{\displaystyle\sum\limits} \)
\( \newcommand{\dint}{\displaystyle\int\limits} \)
\( \newcommand{\dlim}{\displaystyle\lim\limits} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\(\newcommand{\longvect}{\overrightarrow}\)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)Polar Analysis of Functionalized Precursors
Polar reactivity analysis is not very helpful for dislocaton of longifolene because it has no polar functionality. However, functionalized precursors are suggested by considering possible syntheses of the exocyclic methylene. Thus, an alcohol could produce 32 by dehydration, and the alcohol could arise by addition of a methyl nucleophile to ketone 43.

For a direct polar C-C connective synthesis of 43, any additional polar activating functionality in a precursor must be lost during C-C bond formation. Thus, 44-47, precursors for a direct synthesis of 43, result from the four possible disconnections that exploit the potential nucleophilicity of carbon a to a carbonyl, whereas 48 and 49 result from the two possible disconnections that exploit the electrophilicity of a carbonyl carbon. However, it may be advantageous to use an indirect strategy, one that incorporate additional functionality in the penultimate intermediates of skeletal construction (eq. 50 or 51).

That functionality must then be removed after the completion of the carbon network. For example, because the exocyclic methylene of 32 might reasonably be derived from an ester 52, another set of intermediates that has a different reactivity pattern than the first set may be generated by polar reactivity analysis (e.g. 53 and 54). A great many additional precursors may be generated by considering dislocations involving additional activating groups or unsaturation.

Topological Analysis
For molecules like 32, that have minimal functionality and complex skeletons, another approach has been suggested for identifying useful dislocations. Thus, attention is first directed to "an exhaustive analysis of the topological properties of the carbon network to define the range of possible precursors...from which the desired skeleton can be produced by the establishment of one or two connecting bonds."3 Possible reactions, appropriate activating functionality, etc., are only considered after the topological analysis.
In many cases, the most synthetically useful dislocations result from removing one bond between ring-member atoms, called common atoms, that are bonded to three or four other ring members (but not two). For longifolene (32), in which the common atoms are numbered 1-4, this generates three topologically simplified structures 55-57.

Another useful series is generated by removing one bond between a common atom and a noncommon atom. Two members of this series are 58 and 59. Since some reactions generate two new bonds, e.g. Diels-Alder cycloaddition, structures generated by removing two bonds of the original network 32, especially which join two adjacent atoms to one or more common atoms as in 60, should be considered. However, intermediates suggested by dislocations involving removal of a bond between noncommon atoms cannot be disregarded a priori.

After the topological analysis, specific reactions and appropriate functionality to permit bond formation are considered. The process is repeated until a series of potential precursors is generated for each penultimate intermediate and so on until the synthetic tree is complete. As functionality is added to intermediates, topological analysis becomes less relevant. “Maximum utilization of (sub)target-related functionality” (see section 1.2), and hence polar reactivity analysis (see section 1.4), becomes a major factor in synthetic planning. Compounds 61-66 are possible functionalized derivatives corresponding to structures 55-60, respectively.


At some point, a choice between a broad range of possibilities is made. It must necessarily be "very much a function of the methodology of synthetic chemistry available at the time, of certain practical considerations such as the availability of the necessary materials and reagents, and of certain subjective judgments relating to the feasibility of key reactions or the existence of alternatives."3
Fatally Flawed Strategies
To illustrate the pitfalls of designing a complex molecular synthesis, we will first consider some unsuccessful strategies for the synthesis of longifolene. One strategy4 was based on the interconvertibility by rearrangement of longifolene (32) and its hydrochloride 67.

An intermediate 69, related to 65, was prepared in ten steps from D-α-bromocamphor (68), that is readily available from a natural product. However, 69 gave aldol product 71 rather than the desired product 70 under Michael reaction conditions. Thus, the ready availability of the starting material notwithstanding, the ambident electrophilicity of the enone moiety in 69 derailed the synthetic plan.

Removal of a bond between two noncommon atoms in the first dislocation from 32 led to consideration of the potential intermediate 73 for the synthesis of longifolene.5 This route is especially attractive since 73 is easily prepared in a few steps from readily available starting materials. The key cyclization of 73 to 74 failed upon treatment of 73 with acids.

Other modes of cyclization should be examined, such as 73 → 75 → 76. But preferential initial hydroboration of the monosubstituted C=C bond will preclude the required orientation for the addition to the tetrasubstituted C=C bond and lead to 77. Alternatively, an intermediate 79, related to 65, may be available from the Diels-Alder adduct 72 and may undergo intramolecular alkylation delivering 76. Steric approach control should favor the requisite stereochemistry at position 7 in 78.

The first successful synthesis of longifolene (32) involves the key cyclization 61 → 80 as the last step of skeletal construction.3 Incidentally, 61 is suggested not only by topological considerations (i.e. structure 55), but also by polar reactivity analysis (i.e. structure 44). After much experimentation, only a 10-20% yield could be achieved in this crucial step. Conversion of 80 to 32 then involved final addition of a methyl and methylene group and removal of the carbonyl groups.

The synthesis of the key intermediate 61 illustrates a strategy that is useful for carbon skeletal construction, namely ring size modification (RSM). Thus, 61 was prepared from the readily available Wieland-Miescher ketone (81) by expansion of a six to a seven membered ring. The selective ketalization of the saturated carbonyl group in 81 is possible owing to deactivation of the unsaturated carbonyl by the adjacent \(\pi\)-electron system.
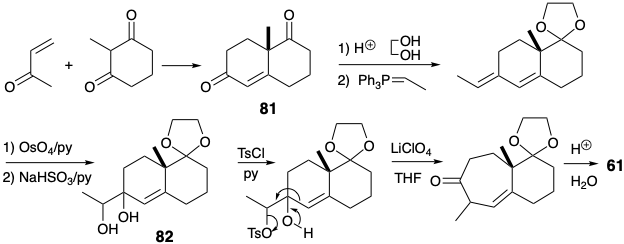
Exposure of diol 82 to the usual acidic conditions for pinacol-pinacolone rearrangement would result in ionization of the tertiary allylic alcohol and produce an acetyldecalin derivative. It was, therefore, necessary to devise a modified procedure to direct the rearrangement of the diol 82 along the desired pathway by facilitating ionization of the secondary hydroxyl. Therefore, the secondary hydroxyl was selectively tosylated. Ionization of the labile tosylate leaving group was accompanied by migration of the vinyl group. The saturated carbon chain is less prone to migrate than the unsaturated one because p- electron participation is possible in the latter but not the former rearrangement.
The Ring Size Modification Tactic
The logic of a synthetic route can be used as a tool for devising a strategy or, ex post facto, as a framework to achieve a fundamental understanding of a known synthesis. The decision to employ ring size modification in the above synthesis of 61 is a logical consequence of topolgical and polar analysis of this target. Topological analysis suggests disconnection of the bicyclic ring system at bonds to the bridgehead carbons which are common atoms. Double disconnection of the seven-membered ring suggests a symmetrical precursor, a 2-substituted 2-methylcyclohexan-1,3-dione. Polar analysis reveals the possibility of a polar annealation for construction of the cyclohexandione that exploits the activation provided by two consonant carbonyl groups. However, one of the desired disconnections of 61 lies on a dissonant circuit. Removal of one atom of this dissonant circuit (ring contraction) produces a consonant circuit in 82 and the possibility of skeletal construction by polar annelation; i.e the Robinson annelation producing 81.

It is important to note that a dissonant circuit in 61 is produced from a dissonant precursor 82. Also, as noted in the previous chapter (see section 3.4), the ring expanding rearrangement of 82 is equivalent to a hypothetical two stage dislocation of the target, disconnection followed by connection. It is also instructive to note the changes in fs that accompany the 82 → 61 rearrangement. Polar disconnection raises f (from +1 to +2) for the electrophilic center undergoing polar disconnection from 82 and lowers f (from +1 to 0) for the electrophilic center undergoing polar connection. The requisite polar reactivity dissonance is created by a nonpolar reaction, oxidative vicinal hydroxylation of an alkene (dioxidative addition). This alkene is obviously derivable from dione 81 by selective Wittig olefination. 81 is entirely consonant. It can be constructed by polar reactions from 2-methylcyclohexan-1,3-dione and methyl vinyl ketone. Had the Robinson annelation process and Wieland-Miescher ketone (81) not been known, the above retrosynthetic analysis would have led to their invention.
Check for Flaws
Having devised the above strategy, it is mandatory to apply step 4 of the "Protocol for Synthetic Design" outlined on page 23. We must examine the strategy for possible flaws. In fact, polar rearrangment of 82 under acid catalysis is expected to follow an alternative pathway involving hydride migration to a tertiary carbenium ion that would be formed more readily than the requisite secondary carbenium ion. Therefore, the strategy was modified to provide selective activation of the secondary hydroxyl. Thus, tosylation enhanced its nucleofugacity.
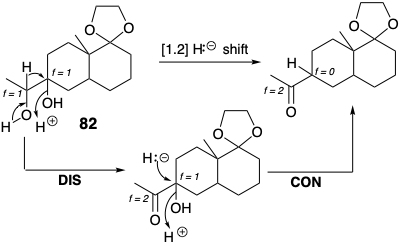
Also note that the concerted hydride migrating rearrangement is equivalent to a two stage dislocation of the target, disconnection followed by connection of H\({}^\ominus\). Furthermore, polar disconnection of hydride raises f (from +1 to +2) for the electrophilic carbon center undergoing polar disconnection (the migration origin) in 82 and lowers f (from +1 to 0) for the electrophilic carbon center undergoing polar connection (migration terminus).
Similar changes in fs accompany polar rearrangements involving nucleophilic carbon at the migration origin and terminus as, for example, in the rearrangement of G3P to DHAP (see section 2.1). This process is actually a two-stage dislocation of the target DHAP: disconnection of H\({}^\oplus\) from C-1 followed by connection of H\({}^\oplus\) at C-2.

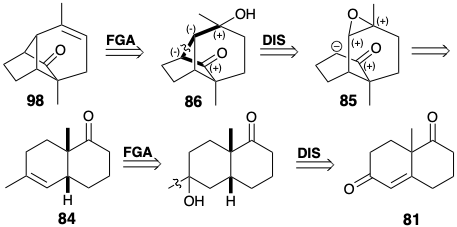
Ring size modification can be applied at any stage of skeletal construction. In the following synthesis, ring expansion is applied after completion of a skeletal network that is topologically equivalent to that in longifolene (32).6 Although the skeletal network in 86 has bridges of different lengths than those in 32, it has the same connectivity as 32. Expansion of one of the bridges in 86 leads to the longifolene ring system (see below). The synthesis of 86 has several important features. As in the previous synthesis of 32, the present appraoch begins with the Wieland-Miescher ketone (81). Catalytic hydrogenation proceeds with stereoselective formation of the cis-decalone 83 owing to steric approach controlled addition of hydrogen to the convex side of the folded ring system of 81. Similarly, epoxidation of 84 occurs with stereoselective delivery of oxygen from the convex side. The stereochemistry of epoxide 85 is ideally suited for nucleophilic attack during intramolecular SN2 alkylation of the corresponding enolate anion. This key cyclization in Mc Murray's longifolene synthesis proceeds in excellent yield (92%).

The longifolene ring system was then generated from 86 by a ring expansion involving pericyclic opening of a cyclopropyl carbenium ion that is generated during solvolysis of 87. The required nucleophilic 1,4-addition of a methyl nucleophile to an enone 88 was accompanied by two undesired reactions. One, the replacement of a vinyl bromo substituent with methyl, generated a useless byproduct. However, the other, an intramolecular aldol condensation, was not a fatal flaw because the extra ring thus formed could be cleaved by a fragmentation reaction (89 → 90).

Ring Expansion as a Three Step Process
Again, let us perform a retrosynthetic analysis ex post facto to achieve a more fundamental understanding of the longifolene synthesis via key intermediates 81-90. We will consider some alternatives that were not adopted, and examine strategic considerations that underlie the pathway that was chosen. In this analysis, we will presume the boundary conditions of using 81 as starting material and generating a tricyclic carbon network by formation of a bond between the incipient common atoms 1 and 2 (numbered as in 55 on page 116) in a bicyclic precursor. Also, functionality will be introduced by presuming a ketone as the progenitor of the exocyclic methylene group. However, instead of forming the tricyclic skeleton at the end of the synthesis after expansion of a 6 to a 7-membered ring, we will first form the tricyclic skeleton and then perform a ring expansion. We could presume that the quaternary carbon bearing the gem dimethyls is inserted into the six-membered ring of a precursor 92 to generate 91. That 91 might contain a second carbonyl adjacent to the bridgehead is the suggested by the fact that this carbon in 92 corresponds to a carbonyl carbon in the starting material 81 (vide infra). The bond to be disconnected between two common atoms in 92 lies on a dissonant circuit between the carbonyls. Therefore, additional functionality, i.e., a nucleofuge, is required in a precursor, X in 93, to allow polar bond formation.

Ring expansion involves insertion of a carbon atom between two ring members. One bond must be formed between the new carbon and each ring member while the bond between ring members must be severed. There are two topologically different ways to accomplish a ring expansion. One possibility for generating 91 from 92 is analogous to the ring expansion of 81 via 82 (see above). Thus, a retro pinacol dislocation of 91 is achieved by disconnecting the bridgehead carbon (as nucleofuge) in 91 from the quaternary carbon and reconnecting it (as nucleophile) to the neighboring carbonyl carbon. This suggests a synthon 94 and synthetic equivalent 95 as precursors of 91. In this strategy, ring expansion is achieved by a connection-disconnecion-connection (CDC) sequence that starts with connection of the nucleophilic carbon of 2-diazopropane to an electrophilic carbonyl carbon of 92.

A topologically different strategy, connection-connection-disconnection (CCD), necessarily involves a cyclopropane intermediate that might be formed by cycloaddition to an alkene 98. Thus, 91 could be derived from a cyclopropane 97 that could isomerize to a cycloheptene precursor 96. Necessarily, only one of the gem methyl groups of 91 can be present in 96 because the carbon bearing this methyl is quaternary in the cyclopropyl precursor 97. Thus, provision must be made for introducing the last methyl group. This might be done by adding functionality to 96, as in 99 that has a carbonyl group conjugated with the carbon center to which a methyl must be added. If the ring expansion that will produce 99 is to involve a polar fragmentation of the ring fusion bond in a cyclopropane intermediate, then retrosynthetic polar analysis suggests two routes to 99. In both routes, the ring fusion bond is provided by retrosynthetic connection to a carbon bearing electropohilic subtarget-related functionality, a carbonyl in 99 or a hydroxyl in 100. The electrons for this connection are provided by an incipient nucleofuge (Nu) through addition to the C=C bond.

The less direct route via 100 is compatible with an alkene precursor 98. Both routes revealed by this analysis involve the cycloadditon of a carbene to which is appended a nucleofuge (Nu). Although departure of the nucleofuge could occur after fragmentation of the ring-fusion bond, alternative timing is possible. The solvolysis of the dibromocyclopropane derived from 98 probably would be a concerted process.
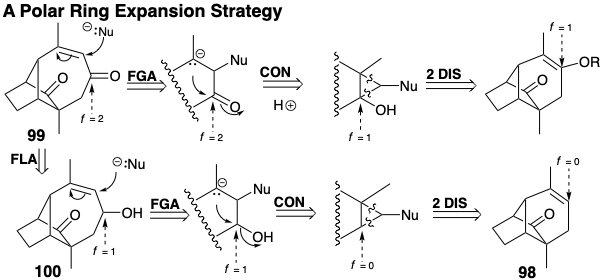
Polar dislocation of 98 to an allylic electrophile and enolate may provide a flawed strategy because the C=C bond in 99 introduces ambident electrophilicity. Thus, cyclization might generate 102 by an SN2' reaction.

Therefore, the C=C bond in 98 is best introduced after cyclization, e.g. by dehydration of alcohol 86. The bond to be disconnected between two common atoms in 86 lies on a dissonant circuit between the carbonyl and hydroxyl groups. Therefore, additional functionality is required in a precursor, e.g. 85, to allow polar bond formation. This epoxide would be obtainable by dioxidative addition to an alkene 84. Generation of 84 from starting ketone 81 is trivial.
Fragmentation of Fused Bicyclics: A Tactic for Generating Larger Rings
During the Mc Murray synthesis of longifolene, an undesired connection formed by intramolecular aldol condensation of enolate 103 generated in the conjugate addition of a methyl nucleophile to intermediate 88. Owing to a proclivity of enolate 105 toward aldol condensation, retro aldol fragmentation of the pentacyclic product 104 could not provide the requisite ring system. This problem was circumvented by an isoelectronic (see page 80) fragmentation after lowering the functionality level of the ketone in 104 to an alcohol. Thus, retro Prins fragmentation of mesylate 89 generated 90 in which the weakly nucleophilic alkene, in contrast with the more strongly nucleophilic enolate in 105, showed no proclivity toward condensation with a carbonyl group.
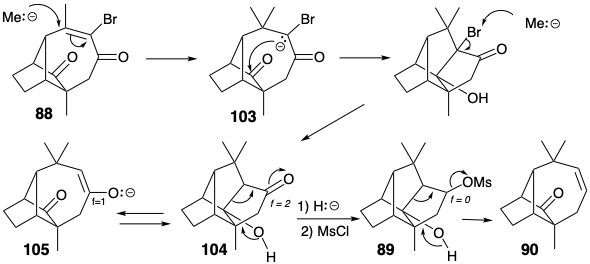
The Mc Murray synthesis of longifolene provides two examples of fragmentation of a bond shared by two fused rings to generate a single larger ring. The first example exploited fragmentation of the cyclopropane 87 as part of a ring expansion tactic, while the second, an unplanned step in the synthesis, involved fragmentation of 89.

Another synthesis of longifolene was designed to exploit the fragmentation of a fused cyclobutane. This strategy recognizes the possibility of using carbonyl functionality in 43 to provide polar reactivity for introducing the methylene and a-methyl groups, and another carbonyl group to allow introduction of the gem dimethyl array into a precursor 106. Dislocation of this subtarget by a polar connection suggests that dione 106 might be generated by the retroaldol fragmentation of a β-hydroxyketone 107. In contrast with the equilibrium between aldol 104 and dione 109 that favors the former, the equilibrium between aldol 107 and dione 106 is expected to favor the latter owing to relief of ring strain associated with cleavage of a cyclobutane.

An exceptionally efficient synthesis of longifolene resulted from the application of this strategy.7a Only 10 steps are used to convert enamine 110 and acyl halide 111 into longifolene in 26% overall yield. Phototolysis of an enol ester derivative of dione 112 folowed by hydrogenolytic removal of the benzyloxycarbonyl (BOC) group generated dione 106 via 107. Selective methylenation of the less sterically congested carbonyl in 106 followed by cyclopropanation, hydrogenolysis of 1 1 3 , and methylation delivered ketone 43, an intermediate in both the Corey and McMurray longifolene syntheses.

A Polyene Cyclization Route
Another efficient synthesis of longifolene (32) is based on a structural simplification suggested by topological analysis. Thus, dislocation to a subtarget 60 (see page 116) by removal of two bonds involving common atoms suggests a precursor containing only one ring. In the synthesis, these two bonds were generated in a key acid-catalyzed polyene cyclization (114 → 115).7b The conversion of 115 to 32 requires reductive removal of the hydroxyl. This was accomplished by an SN1 replacement of hydroxyl by hydride through an intermediate carbenium ion 116. To provide polar activation that could be exploited for introducing the angular methyl group, the C=C bond in 117 was isomerized to an exocyclic methylene in 118. Oxidative cleavage then delivered ketone 119.

A synthesis of 114 from a methylenecyclopentanone electrophile 120 and nucleophilic side chain synthon 121 is suggested by polar analysis. Two extra steps were added to the synthesis to allow purification of the enolate 122 produced by the 1,4-addition of 120 to 121. Thus, 122 was trapped by O-acylation. After purification of the enol acetate 123, the enolate 122 was regenerated and then brominated. Dehydrobromination and reduction completed the synthesis of 114.
